

Castleman disease (CD) is a rare lymphoproliferative disorder that can present in different ways. It was first described by Benjamin Castleman in 1954 in a case report that identified a symptomatic solitary lymph node showing hyaline vascular histopathological changes [1]. Over the years, we have recognised, both through clinical behaviour and pathological features, many variants of CD. The broadest classification is divided into unicentric CD (UCD) (localised disease) and multicentric CD (MCD) (generalised disease). When either UCD or MCD is suspected, preferably an excision biopsy of a lymph node is undertaken to determine the histopathological variant of CD [2]. Figure 1 summarises the diagnostic process.
Figure 1
The diagnostic process of Castleman disease.
CD – Castleman disease, EBV – Epstein-Barr virus, HHV-8-associated MCD – human herpesvirus 8-associated multicentric Castleman disease, HIV – human immunodeficiency virus, HLH-MAS – hemophagocytic lymphohistiocytosis/macrophage activation syndrome, IgG4 – immunoglobulin G4 related disease, iMCD – idiopathic multicentric Castleman disease, MCD – multicentric Castleman disease, POEMS-MCD – peripheral sensorimotor neuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin lesionsassociated multicentric Castleman disease, RA – rheumatoid arthritis, SLE – systemic lupus erythematosus.
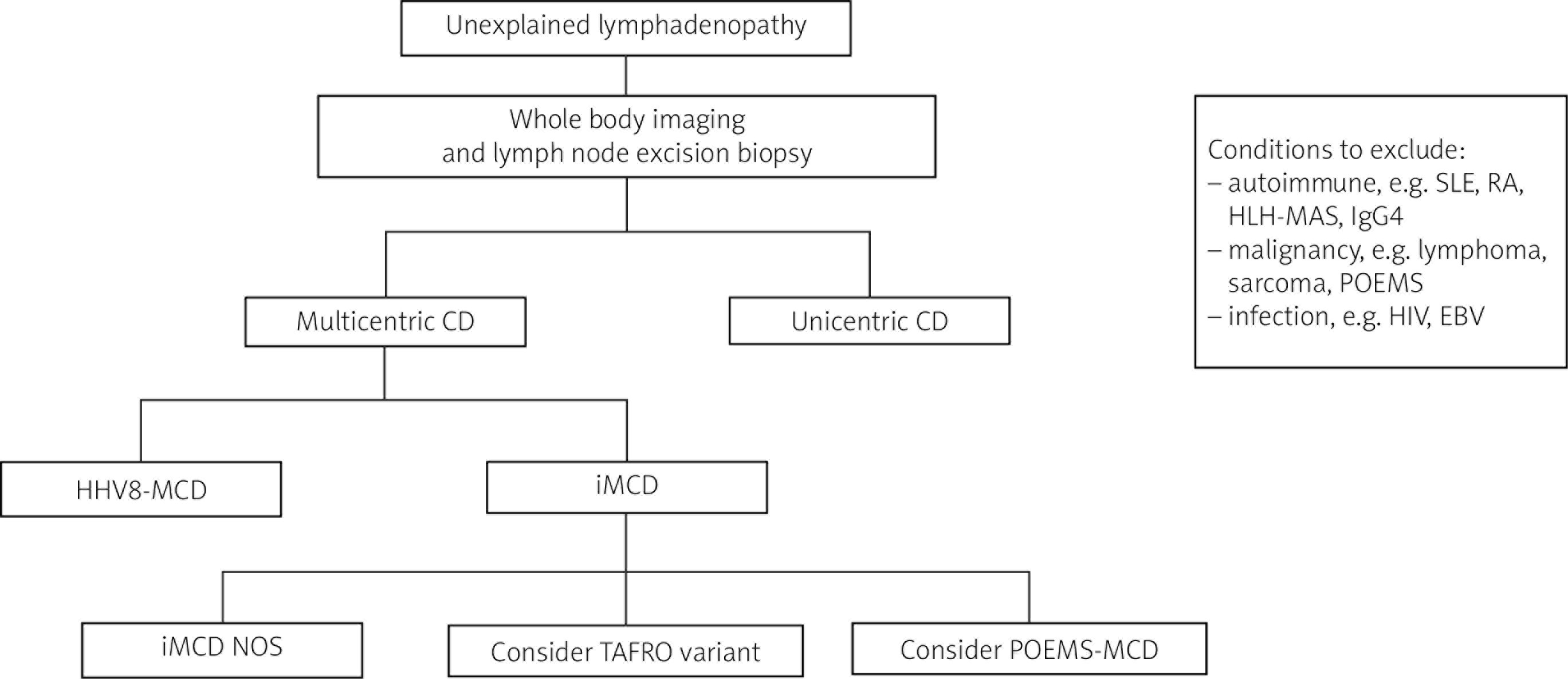
Unicentric CD presents with localised lymphadenopathy, often in the neck or chest [2]. Whole-body imaging (computed tomography, magnetic resonance imaging, or positron emission tomography) can help determine if one or more lymph node groups are affected and if there are other lymph node regions present [3]. This helps to differentiate between UCD or MCD.
The most common histologic variant in UCD is “hypervascular”. With this subtype, if symptoms are present, they are often mild, localised, and related to a mass organ effect [3]. Systemic symptoms are associated with excess interleukin-6 (IL-6) secretion, which is more commonly associated with the “plasmacytic” variant (10–25% of cases of UCD) [3]. Patients with the plasmacytic variant are more likely to have B-symptoms, anaemia, and hypergammaglobulinaemia when compared to other variants [4].
Treatment may not be necessary if the patient is asymptomatic, and a period of observation can be considered [2]. For UCD, complete surgical excision of the node/nodal group is often curative and is preferred [2, 3, 5]. If the affected nodes are not amenable to total resection, then partial resection or debulking with treatments such as radiotherapy or weekly 375 mg/m2 rituximab infusions can be considered [2]. The prognosis of UCD patients who have treatment is excellent, particularly with total surgical resection [3, 5]. Dispenzieri et al. [4] reported a 5-year overall survival rate of 91% for UCD patients, which included patients who were observed rather than undergoing treatment.
Multicentric CD presents with systemic lymphadenopathy with associated constitutional symptoms. It can be subdivided into human herpesvirus 8-associated MCD (HHV-8-associated MCD), peripheral sensorimotor neuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin lesions (POEMS)-associated MCD (POEMS-MCD), or idiopathic MCD (iMCD). As with UCD, an excisional lymph node biopsy is required to determine the underlying histologic variant.
Human herpesvirus 8-associated MCD is commonly associated with human immunodeficiency virus (HIV). Clinical datasets show that patients with HIV and MCD have HHV8-associated MCD. However, there are occasional patients with HHV8-associated MCD who are HIV negative [6]. Human herpesvirus 8-associated MCD is driven by the secretion of a virally derived analogue of IL-6 [2, 6]. Anti-retroviral therapy should be commenced/continued for all patients with a diagnosis of HIV. Treatment with rituximab has been shown to increase overall survival from 33% to 90% at 5 years [8]. Rituximab with etoposide may be considered for those with more severe disease or those with a poor performance status [2].
Peripheral sensorimotor neuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin lesions-associated MCD is rare and can be difficult to diagnose. It is associated with either a monoclonal plasma cell disorder (usually IgA or IgG with λ light-chain restriction) or the presence of sclerotic or lytic bone lesions [2, 7]. The treatment of POEMS-MCD varies from other variants; high-dose chemotherapy followed by autologous stem cell transplant can be effective and may be recommended by specialist multi-disciplinary teams with prior experience in the diagnosis and treatment of this rare disease [2, 9].
Idiopathic MCD is challenging to diagnose; it can be reliably diagnosed when HHV8-associated MCD, POEMS-MCD, and CD-mimics (certain infections, autoimmune conditions, and malignancies) have been excluded [2, 7]. Idiopathic MCD is driven by systemic inflammation caused by cytokine release, in particular IL-6. Clinical features include systemic lymphadenopathy, B-symptoms, cytopaenia, polyclonal gammopathy, and organ dysfunction (sometimes multiple organs), and it can range from mild to severe.
A more severe variant of iMCD is TAFRO, which is characterised by thrombocytopaenia, anasarca, fever, reticulin myelofibrosis, and organomegaly.
The histologic variants of iMCD are “hypervascular”, “plasmacytic”, and “mixed”, with TAFRO variant patients often showing a mixed pathology [7].
In Europe and the USA, the only licenced treatment of iMCD is siltuximab. Siltuximab is a monoclonal antibody that prevents the binding of IL-6 to soluble and membrane-bound IL-6 receptors by forming high-affinity complexes with IL-6 [10]. In the licensing trial, there was an overall response of 51% for those treated with siltuximab vs. 0% for placebo at 14-month follow-up [11]. Subsequent real-world data showed that treatment with siltuximab achieved a complete response in 81.5% of participants [12], and a durable symptomatic response was achieved at a median of 23.5 days after the initiation of siltuximab treatment [12]. UK real-world data show that 72% of participants achieve a biochemical response within 3 months of treatment initiation, and that the treatment is well tolerated [13].
In summary, CD is a rare disease with varying clinical presentations that may mimic other diseases. Whole body imaging and excisional lymph node biopsy are key to establishing a diagnosis of CD and the subtype. A diagnostic criteria has been published by an international working group in 2017 [7] to aid clinicians with the diagnosis of iMCD [7]. Due to the complexity of CD, early input from a specialist with experience of this disease is critical.