
Introduction
Deficiency of adenosine deaminase 2 (DADA2) is an autosomal recessive and auto-inflammatory disorder. It is a monogenic disease that is caused by loss-of-function variants in the ADA2 gene (formerly known as the CECR1 gene), which is located on chromosome 22q11.1. Deficiency of adenosine deaminase 2 was first described in 2014 by two separate studies by Navon Elkan et al. [1] and Zhou et al. [2].
Deficiency of adenosine deaminase 2 is a disorder with high variability of age at onset, disease findings, severity, clinical courses, and organ involvement. This variability may even occur among family members or those with the same mutations. Deficiency of adenosine deaminase 2 has a wide spectrum of clinical presentations which range from fatal systemic vasculitis with multiple strokes (in pediatric patients) to limited cutaneous disease (in middle-aged patients).
The clinical features of DADA2 may include polyarteritis nodosa (PAN)-like features such as livedoid rash, early-onset stroke, hematological abnormalities (such as cytopenia), hypogammaglobulinemia, and systemic inflammation [3–7].
The first-line treatment of choice in DADA2 is tumor necrosis factor α (TNF-α) inhibitors, which control inflammation and preserve vascular integrity [3, 8–10].
Since DADA2 is a disorder with extensive variability in genotypic and phenotypic features and allele frequency of ADA2 mutation variants, the prevalence of DADA2 may be higher than its current estimate, especially in certain populations.
Therefore, screening the ADA2 gene in pediatric patients who present with a rash, vasculitis, PAN, early-onset neurological features, and systemic inflammation may help to early diagnose DADA2. A clearer knowledge of the wide range spectrum of DADA2 presentations would help to better diagnose and manage the patients, which would consequently improve their clinical outcome [8, 11].
We aimed to provide an overview of the known pathophysiology, clinical presentations, diagnosis, and treatment of DADA2 to help clinicians to better diagnose and treat these patients. As a result, with early prevention and decrease of complications, DADA2 patients may experience a better quality of life.
Material and methods
We searched the PubMed/Medline, Web of Science, Scopus, and Google Scholar databases over the period 2000–2021 for the relevant literature using the following key words: deficiency of adenosine deaminase 2, DADA2, ADA2 gene, CECR1 gene, early-onset stroke, early-onset vasculopathy, ADA2 deficient PAN, and early-onset PAN.
Epidemiology and demographics
Deficiency of adenosine deaminase 2 is a rare disease with about 200 reported cases since its first description in 2014. Consequently, most mutational variants of ADA2 are novel or rare (allele frequency of 1 : 1000). It has also been reported that the prevalence of DADA2 is higher in populations with high parental consanguinity.
In addition, some specific variants are more common in certain populations. For example, p.Gly47Arg and p.Arg169Gln are more frequent in Georgian-Jewish and Northern European populations, respectively [4].
Since DADA2 and PAN have many similar clinical features, DADA2 may be initially misdiagnosed as PAN [12]. It has been estimated that DADA2 has a prevalence of 4 per 100,000 individuals [13].
Pathophysiology
The extracellular enzyme adenosine deaminase 2 (ADA2) is encoded by the ADA2 gene and is mainly expressed by monocytes/cells of the myeloid lineage [14]. The ADA2 enzyme has different effects. It is a growth factor that is involved in the development of endothelial and hematopoietic cells, has autocrine activity, promotes the proliferation of monocytes, and induces the differentiation of macrophages. Patients with deficiency of adenosine deaminase 2 have defective differentiation of M2 macrophages (anti-inflammatory effects) [15].
Consequently, DADA2 leads to increased M1 macrophages that release pro-inflammatory cytokines from monocytes and macrophages. These cytokines induce inflammation, damage the endothelial cells, and injure the vessel walls [14–16].
Therefore, loss-of-function mutations in the ADA2 gene disrupt the integrity of the vessel wall by decreasing the activity of the ADA2 enzyme. Some of the clinical features of DADA2 are associated with small or medium-sized vessel vasculitis. For example, DADA2 has been associated with lymphopenia, hypogammaglobulinemia, and systemic vasculopathy [1–3, 5, 6, 8, 14, 15].
In addition, it has been recently suggested that the interferon (IFN) pathway has a role in DADA2 since increased IFN (type I) signatures have been detected in DADA2 [13]. Furthermore, ADA2 is an enzyme that converts adenosine and 2′-deoxyadenosine to inosine and 2′-deoxyinosine, respectively. Deficiency of adenosine deaminase 2 regulates the activation of neutrophils as neutrophils express adenosine receptors. Decreased ADA2 activity may cause endothelial damage by chronic and increased activation of neutrophils.
In addition, myeloperoxidase (MPO) has high levels in neutrophils and has been associated with the dysfunction of endothelial cells. Consequently, the ADA2 enzyme may cause endothelial damage through its roles as the regulator of neutrophil activation and an inhibitor of MOP expression [10].
A study in Japan showed that both types of INF (type I and type II) signaling pathways were upregulated in DADA2. They also reported that the STAT1 gene may be involved in the pathogenesis of DADA2 [17].
Human et al. [9] summarized the potential pathogenesis of DADA2 in three main pathways. First, DADA2 causes growth factor dysfunction leading to impairment of the endothelial integrity and development and, in turn, vasculopathy. Secondly, it causes immune dysfunction resulting in disrupted neutrophil activation and subsequent vasculopathy.
In addition, dysregulation of macrophages/monocytes occurs, which leads to increased production of pro-inflammatory cells. The immune dysfunction also causes mild B cell dysfunction and subsequent mild immunodeficiency. Finally, DADA2 causes catalytic dysfunction that leads to increased levels of adenosine. As a result, inflammation, tissue damage, and fibrosis occur [9].
Furthermore, neutrophils and low-density granulocytes (LDGs) (a subset of neutrophils) form neutrophil extracellular traps (NETs) and are involved in the pathogenesis of vasculitis [18].
Carmona-Rivera et al. [18] found that NET formation by neutrophils (NETosis) may be the cause of macrophage activation leading to TNF-α production in DADA2 [19]. They reported that NETs and macrophages were present in the affected GI tissue of DADA2 patients (in vivo).
They also reported that there were increased levels of circulating LDGs during the active phase of DADA2 which decreased following treatment with TNF-α inhibitors, suggesting a link between inflammation and NET-osis in DADA2 [18].
In addition, adenosine triggered NETosis by interacting with A1 and A3 adenosine receptors (through reactive oxygen species [ROS] and peptidylarginine deiminase [PAD]). Furthermore, M1 macrophages and NETs from DADA2 patients significantly released TNF-α, which indicates the association between ADA2 dysfunction and inflammatory cytokine production [18].
The results of this study demonstrated the pathogenic role of NETosis in DADA2. The authors also concluded that modulating the NET formation which is induced by adenosine could have therapeutic potential in these patients [18].
Genetic causes
Deficiency of adenosine deaminase 2 is an autosomal recessive monogenic disorder and the majority of the patients have loss-of-function variants in the coding region of ADA2. Pathogenic mutations can occur in all 4 domains of the ADA2 protein and have been found in every exon of the gene.
Most DADA2 cases have bi-allelic mutations in the ADA2 gene and the majority of the mutations are missense mutations (about 80%). Other variations include copy number variations (CNVs) including single or multiple exons and frameshift, insertions/deletions, splicing, and nonsense mutations [4, 15, 20].
To date, 126 mutations of the ADA2 gene have been reported on the online database “Infevers” (The registry of Hereditary Auto-inflammatory Disorders Mutations). These mutations include 100 substitutions (79.36%), 20 deletions (15.87%), 2 del-ins (deletion-insertions) (1.58%), 3 duplications (2.38%), and 1 insertion (0.79%) [21].
However, there have been reports of DADA2 with typical clinical and enzymatic phenotypes without any mutations [4, 15]. For example, in 2017, Caorsi et al. [15] reported several cases of patients who presented with typical clinical features of DADA2 such as vasculopathy, stroke, and hypogammaglobulinemia. However, these patients had non-confirming or negative genetic analyses [15].
This suggests that some DADA2 mutations may include null alleles, intronic mutations, noncoding regulatory areas, or insertions/deletions [4, 9, 15]. Half of the DADA2 patients have homozygous mutations. The most common mutation is reported in Georgian-Jewish and Turkish populations (G47R mutation). This is because of the increased carrier frequency. This mutation is also seen in South Asia. G47R mutations are homozygous and present with a classic PAN-like phenotype.
On the other hand, the p.Arg169Gln (R169Q) mutation is usually observed in the Northern European population [1, 4, 9, 11, 15, 20, 22].
Genotype-phenotype
Deficiency of adenosine deaminase 2 is a disorder that has a wide spectrum of clinical findings and involves multiple organs. It also has heterogeneity, which is even observed among family members with similar mutations [23].
The phenotypes of DADA2 include PAN-like vasculitis, hematological features, immunodeficiency, autoinflammation, and lymphoproliferation. Many patients have overlapping presentations. However, some patients may be asymptomatic or have mild symptoms that are diagnosed in family screening [24].
Patients who are homozygous for the same founder mutation variant or have the same biallelic heterozygous mutation variant present at varying ages, frequency, and severity [4]. For example, patients who have a homozygous R169Q mutation have variable presentations (vasculitis, hematologic abnormalities, or immunodeficiency). Studies have tried to establish a genotype-phenotype association. Some were not able to find such an association [1, 4, 9, 11, 15, 20, 22].
However, Özen et al. [22] compared the PAN-like and DBA/immunodeficiency phenotypes with similar ADA2 levels and reported a possible genotype-phenotype association. They reported that mutations of the dimerization domain were associated with the PAN-like phenotype and mutations of the catalytic domain were associated with hematological features [22].
In addition, another study compared the DADA2 genotypes with phenotypes and levels of ADA2. They reported that mutations that caused minimal residual ADA2 activity were associated with hematological phenotypes (such as pure red cell aplasia and DBA) and mutations with at least 3% enzymatic activity were associated with the PAN-phenotype [25].
Furthermore, Lee et al. [25] reported that in DADA2, vasculitis and strokes are associated with hypomorphic missense mutations that cause low ADA2 protein activity, and mutations that cause a complete lack of ADA2 protein are associated with hematological presentations.
On the other hand, it has been reported that the amount of activity of the remaining ADA2 enzyme may be more predictive of DADA2 phenotypes compared to the genotypes [23]. It has also been suggested that since patients with identical mutations have diverse clinical features and severity, it is likely that environmental factors, genetics, and epigenetic modifications are involved in DADA2 [20].
Further investigations on DADA2 will broaden the knowledge about the genotype-phenotype associations [7].
Clinical presentations
Many of the clinical features of DADA2, knowledge of which is expanding with the ongoing reports, are the result of its involvement in the small- and medium-sized vessels [5]. Deficiency of adenosine deaminase 2 presents with a wide range of clinical manifestations ranging from mild skin features to severe organ involvement that may be lethal.
For example, in pediatric patients, there may be fatal systemic vasculitis with multiple strokes, and in middle-aged patients, there may be a limited cutaneous disease. The common clinical features of DADA2 are presented in Table I.
Table I
Summary of common clinical features of deficiency of adenosine deaminase 2 [1, 2, 4–7, 9, 10, 13–15, 20, 26, 27]
Constitutional symptoms
The common constitutional symptoms include recurrent fever, myalgia, and arthralgia.
Vasculitis and vasculopathy manifestations
Skin manifestations
The common cutaneous features include livedo racemose, livedoid rash, livedo reticularis, maculopapular rashes, PAN nodules, Raynaud’s phenomenon, digital gangrene ulcers, and verrucae vulgaris (warts).
Nervous system
The common clinical manifestations of the nervous system include stroke (ischemic, hemorrhagic, lacunar, ventricular hemorrhage), transient ischemic attack, central neuropathy, peripheral neuropathy, permanent polyneuropathy, cranial nerve palsy, transverse myelitis, transient mononeuritis, optic neuritis, optic nerve atrophy, ptosis (cranial nerve [CN] III palsy), external ocular muscle palsies (diplopia, strabismus), uveitis, occlusive retinal vasculitis (monocular or binocular permanent vision loss), unilateral amaurosis fugax attacks, and sensorineural hearing loss.
Pulmonary and cardiovascular manifestations
The pulmonary and cardiovascular manifestations in DADA2 include necrotizing pneumonia, systemic hypertension, and cardiomyopathy. Sahin et al. [5] reported that although hypertension may occur in DADA2, malignant hypertension is not common.
Gastrointestinal manifestations
The common gastrointestinal (GI) presentations include mesenteric vasculitis and ischemia (recurrent abdominal pain, diarrhea, hepatosplenomegaly, pancreatitis, portal hypertension, hematochezia, bowel perforation, massive bleeding), weight loss, chronic gastritis, bowel stenosis, hepatomegaly, splenomegaly, hepatosplenomegaly, elevated hepatic transient elastography, portal hypertension, and splenic infarcts.
Renal manifestations
The renal features of DADA2 include nephrogenic hypertension, focal glomerulosclerosis, renal amyloidosis, and renal cortical lesions.
Hematologic, immune, and inflammatory abnormalities
Patients with deficiency of adenosine deaminase 2 may present with various hematologic and immune abnormalities, including diffuse adenopathy, abnormal serum Igs, hypogammaglobulinemia, low IgG, low IgM, low IgA, abnormal lymphocyte phenotyping, low class-switched memory B cells, low memory T cells, low NK cells, cytopenia, pancytopenia, neutropenia, leucopenia, lymphopenia, thrombocytopenia, neutrophilic leukocytosis, decreased levels of hemoglobin (Hb), severe anemia, pure red cell aplasia, Diamond-Blackfan anemia (DBA)-like phenotype (pure erythrocytic anemia), lymphoproliferative diseases, elevated estimated sedimentation rate (ESR), elevated C-reactive protein (CRP), low serum iron, positive antinuclear antibody (ANA), positive anti-neutrophil cytoplasmic antibodies (ANCA), and inadequate specific antibody testing.
Other manifestations
Kaya Akca et al. [13] reported that DADA2 patients had increased levels of TNF-α, tyrosine-protein kinase receptor-1 (Tie-1), soluble vascular endothelial growth factor receptor-1 (sFlt-1), soluble isoform of a receptor for advanced glycation end-products sRAGE, Tie-2, and interleukin-18 (IL-18).
The higher levels of IL-18 in DADA2 were associated with hematological features. The level of IL-18 or IFN-γ is increased in the DBA-like phenotype. The increased levels of Tie-1 and IL-18 levels were higher in DADA2 with immunological findings. In addition, the serum levels of sRAGE were higher in DADA2 with neuropathy compared to DADA2 without neuropathy and healthy controls [13].
Diagnosis
Enzymatic and genetic testing
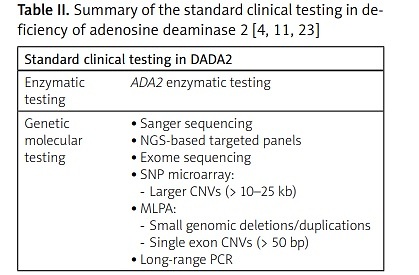
Enzymatic testing in addition to genetic testing is the standard clinical testing for diagnosing DADA2 [4]. Deficiency of adenosine deaminase 2 enzymatic testing of less than 5% of normal or undetectable ADA2 activity confirms the diagnosis of DADA2. The genetic molecular tests for DADA2 include Sanger sequencing, next-generation sequencing (NGS), exome sequencing, single nucleotide polymorphism (SNP) microarray, multiplex ligation-dependent probe amplification (MLPA), and long-range polymerase chain reaction (PCR).
Single nucleotide polymorphism microarray is used to detect larger CNVs of > 10–25 kb, and MLPA analysis may be used to detect small genomic deletions/duplications or single exon CNVs of > 50 bp (Table II).
Table II
| Standard clinical testing in DADA2 | |
|---|---|
| Enzymatic testing | ADA2 enzymatic testing |
| Genetic molecular testing | |
It has also been suggested that measuring ADA2 activity before genetic testing would be more cost-beneficial [3, 20].
Imaging and angiography
Computed tomography scans and conventional angiography are not effective in detecting small lacunar strokes. Magnetic resonance imaging (MRI) is the most suitable imaging modality in detecting cerebral strokes. However, conventional angiography may be useful in detecting aneurism and stenosis in medium-sized vessels [6].
Treatment
Tumor necrosis factor α inhibitors
Tumor necrosis factor α inhibitors (such as etanercept, infliximab, and adalimumab) are considered as the first-line treatment in DADA2 [8, 15]. Early diagnosis and therapy with TNF-α inhibitors are important in preventing severe complications in DADA2 [15].
Tumor necrosis factor α inhibitors have many benefits in these patients, including inflammation control and decrease in inflammatory markers (such as ESR and CRP), maintenance of vascular integrity, stroke prevention, and inhibition of TNF-α production by M1-type macrophages [7, 28–30].
In addition, there have been reports of an increase in the serum levels of iron and improvements in white blood cell (WBC), hemoglobin, hematocrit, and platelet counts. Moreover, the patients have shown improvements in hepatic findings (both vascular and parenchymal) and erythema nodosum and nodules (caused by vasculitis).
There has also been a subjective improvement of livedo racemosa in some DADA2 patients. However, DADA2 patients with hematologic and immune abnormalities have shown varied responses to TNF-α inhibitors [7, 29].
Treatment with TNF-α inhibitors is usually efficient in patients with the vascular type of DADA2. However, it is insufficient in patients with severe neutropenia or pancytopenia. These patients may benefit from other treatment options such as hematopoietic cell transplantation (HCT) [7].
Other treatment options
Caorsi et al. [15] reported that thalidomide might be a less expensive but effective drug as a first-line treatment drug and alternative to TNF-α inhibitors. Other treatment options for DADA2 may include glucocorticosteroids or cytokine inhibitors [4, 31, 32]. Glucocorticosteroids used to be the main treatment option in patients who presented with early-onset PAN and recurrent strokes.
However, there were variable outcomes in these patients and some developed flares of inflammation and vasculitis following the tapering of glucocorticosteroids. In addition, other drugs such as azathioprine and methotrexate have not been proved to be effective in these patients as well [1, 2, 8].
Schnappauf et al. [4] reported that those DADA2 patients who had two null variants and no ADA2 activity are likely to be resistant to treatment with TNF-α inhibitors. Therefore, these patients may be considered for transplantation [4].
Treatment with enzyme replacement therapy in late-onset DADA2 and clinical presentations of combined immunodeficiency has been reported to be effective [33]. There is controversy over prophylactic therapy with anti-platelet and anticoagulant medications for stroke due to the risk of developing hemorrhagic events [34].
In addition, it has been reported that exogenous replacement of ADA2 through fresh frozen plasma causes a transient increase in serum ADA2 levels. Therefore, treatment with fresh frozen plasma does not seem to be an option in treating DADA2 [30].
Hematopoietic cell transplantation
The first international study on the outcome of the hematopoietic cell transplantation (HCT) in DADA2 was conducted by Hashesm et al. [34]. They found that HCT treated the immunological, hematological, and vascular phenotypes of DADA2 and suggested HCT as an effective treatment for DADA2 [32].
Other studies have also reported similar results on the efficacy of HCT in reversing refractory cytopenia, vasculopathy, and immunodeficiency [35]. Deficiency of adenosine deaminase 2 with severe neutropenia or pancytopenia is usually treated with HCT. However, if HCT is not an option, other treatment options such as aggressive suppression of T cells may be considered [7].
On the other hand, HCT is associated with some adverse effects, has limited availability of human leukocyte antigen (HLA)-matched donor, and causes morbidity. Therefore, finding an alternative treatment option seems necessary [29, 36].
Gene therapy
Gene therapy is a potential treatment option for DADA2 and research regarding gene therapy is still ongoing [7]. For example, Zoccolillo et al. [37] investigated the possibility of curing DADA2 with genetic correction through autologous hematopoietic stem and progenitor cells (HSPCs).
They concluded that lentiviral-mediated ADA2 gene transfer corrects the ADA2 enzyme deficiency in the macrophages of DADA2 patients and subsequently prevents excessive inflammation [37].
In addition, Khon et al. [38] reported successful treatment of severe combined immunodeficiency due to ADA deficiency (ADA-SCID) with ex vivo lentiviral HSPC gene therapy [38].
A summary of the treatment options in DADA2 is shown in Table III.
Screening
Screening for ADA2 mutations has been recommended in patients with signs and symptoms of vasculopathy (similar to PAN), particularly in cases with early-onset clinical features or a history of strokes, inflammatory diseases, and family history [5, 15].
In addition, DADA2 should be considered in patients with early-onset PAN-like features who have consanguineous parents due to its autosomal recessive inheritance nature [3].
Early diagnosis and therapy with TNF-α inhibitors are important in preventing severe complications [15] as some of these complications could be potentially life-threatening but might be treatable [3].
Frequent neurodevelopmental evaluation such as evaluation of hearing impairment is recommended in DADA2 [33]. Furthermore, Schnappauf et al. [4] recommended a combination of genetic and enzymatic testing in asymptomatic family members of DADA2 patients to detect individuals at risk [4].
Prevention
There is controversy over treating asymptomatic DADA2 with TNF-α inhibitors. Some studies argue that treatment with TNF-α inhibitors would put asymptomatic patients at risk of side effects such as immune suppression.
In addition, there is concern about the decrease in the efficacy of TNF-α inhibitors following long-term use [39]. Other researchers believe that treatment of all DADA2 patients without contraindications seems necessary as there is still insufficient knowledge about the risk factors that cause stroke in these patients [7].
For example, Schnappauf et al. [4] recommended prophylactic treatment of asymptomatic DADA2 [4].
Prognosis
The exact clinical outcome of DADA2 is still unknown and it has a wide spectrum of severity from mild to lethal [6]. Involvement of the central nervous system, strokes, and the GI system are common causes of morbidity in DADA2 and are associated with poor prognosis [12, 26].
The majority of DADA2 patients present before the age of 10 years and approximately 25% of the patients present before the age of one year. About 8% of DADA2 patients die at a young age (under 30 years). The majority of the patients die due to complications caused by recurrent stroke or infection [8, 40].
Deficiency of adenosine deaminase 2 and polyarteritis nodosa
Since DADA2 and PAN have many similar clinical features, DADA2 may be initially misdiagnosed as PAN. It has been reported that about 25% of patients with DADA2 have been misdiagnosed with childhood PAN [20].
The common clinical characteristics between these two disorders are due to the involvement of small- and medium-sized vessels. For example, the recurrent clinical features of PAN, such as angiographic findings, hypertension, increased acute phase reactants, myalgia, and polyneuropathy, also occur in DADA2. In addition, patients with both disorders may have a positive family history or a positive history of parental consanguinity [5, 6, 15, 26].
However, Huang et al. [20] reported that some features may help to distinguish DADA2 from PAN (Table IV).
Table IV
Some differences between polyarteritis nodosa (PAN) and deficiency of adenosine deaminase 2 [13, 33]
| PAN | DADA2 |
[i] CNS – central nervous system, DADA2 – deficiency of adenosine deaminase 2, Ig – immunoglobulin, PAN – polyarteritis nodosa, PNS – peripheral nervous system, sFlt-1 – soluble vascular endothelial growth factor receptor-1, sRAGE – soluble isoform of a receptor for advanced glycation end-products, Tie-1 – tyrosine-protein kinase receptor-1, TNF-α – tumor necrosis factor α, WBCs – white blood cells.
For example, DADA2 usually presents earlier and at younger ages. Skin manifestations are more commonly seen in childhood PAN and DADA2 compared to adult PAN. The livedo presentation of the skin is more common in DADA2 than PAN. The peripheral nervous system (PNS) is more involved in adult PAN.
Central nervous system involvements such as ischemic stroke and brain bleeding occur more in DADA2 (Table IV). Polyarteritis nodosa patients usually have increased WBC and platelet counts. However, DADA2 patients usually have decreased immunoglobulin (IgA, IgM, and IgG) levels and decreased platelet and WBC counts [20, 22, 23, 27, 29, 41–43].
Furthermore, Kaya Akca et al. [13] reported that the levels of TNF-α, Tie-1, and sFlt-1 were higher in DADA2 patients compared to healthy controls and PAN patients.
In addition, DADA2 patients had higher levels of sRAGE and Tie-2 compared to PAN patients (Table IV). The serum levels of IL-18 were high in both disorders [13].
Conclusions
Deficiency of adenosine deaminase 2 is an autosomal recessive disease that is caused by loss-of-function variants in the ADA2 gene. It has a wide spectrum of clinical presentations and the phenotypes have expanded ever since its first description in 2014. Early diagnosis and treatment of DADA2 are crucial as the clinical features could be potentially life-threatening but might be treatable.
Moreover, a clearer knowledge of DADA2 may help to better diagnose, manage, and improve the clinical outcome of DADA2.
Further studies are also required to investigate the genotype-phenotype associations, the exact pathophysiology, and other therapeutic options.
In addition, there are other aspects of DADA2 that are still not yet fully understood and should be considered in future studies such as its genetics, activity of the ADA2 enzyme, and other possibly involved factors.