
Introduction
Idiopathic inflammatory myopathies (IIMs) are a group of systemic connective tissue diseases that present with muscular and extra-muscular manifestations, diagnosed mostly with the presence of skeletal muscle weakness, elevated levels of muscle enzymes, and the myopathy pattern of electromyography along with characteristic histopathological changes on muscle and/or skin biopsy. There are antibodies, both associated with, and specific for, myositis, and magnetic resonance imaging (MRI) of muscles, which are increasingly used to support the diagnosis [1].
Nailfold capillaroscopy (NFC) is also a non-invasive imaging technique that allows direct observation of microangiopathic changes of the nailfold area; multiple studies have shown its diagnostic and prognostic role in some IIMs, especially dermatomyositis (DM) [2].
Based on the EULAR/ACR classification criteria for adult and juvenile IIMs and their major subgroups, the main groups are polymyositis (PM), DM, inclusion body myositis (IBM), immune-mediated necrotizing myopathy (IMNM), amyopathic dermatomyositis (ADM), and juvenile dermatomyositis (JDM) [3].
The dermatomyositis subgroups have some characteristic skin manifestations, and over the past decade, new subgroups for this disease have been described based on clinical and some myositis-specific antibodies [4].
Although there are multiple kidney manifestations reported in systemic connective tissue diseases, especially in systemic lupus erythematosus (SLE), Sjögren’s syndrome, mixed connective tissue disease, and scleroderma, there are few reports on kidney involvement as an extra-muscular manifestation of patients with IIMs, especially in DM [5].
Although some articles have reported a prevalence of 21–23%, it is mostly in the form of a chronic autoimmune-based glomerulonephritis (GN) or an acute rhabdo-myolysis-induced acute kidney injury (AKI) [6].
The patients in the former group in some studies had an overlap with SLE or other connective tissue diseases [7]; in pure forms of PM, they usually presented with a picture of mesangial proliferative GN, while DM patients more often showed membranous nephropathy.
The second type of kidney manifestations was mostly seen in patients with cancer-associated myositis and was not a direct manifestation of IIM; rather, it was due to post chemotherapy, drug toxicity, or they had co-incidence of sepsis, dehydration, and electrolyte imbalances [6].
Therefore, we aimed to evaluate the clinical, laboratory, capillaroscopy, and pathologic manifestations of patients with DM, who presented with proteinuria and active GN during their first year of clinical manifestation.
Material and methods
In this study, we evaluated the profile of 205 patients who were referred from rheumatology clinics and in-hospital wards of Shiraz University of Medical Science, including Motahary/Namazee, Faghihi, and Hafez, with proximal muscle weakness or high muscle enzymes to the nailfold capillaroscopy clinic of Hafez Hospital from April 2010 to October 2021.
The profiles of patients who met the inclusion criteria for DM with a duration of ≤ 12 months from the first manifestation were re-evaluated by the rheumatologist involved in this study using their referral data and admission notes in hospitals or clinics for the presence of proteinuria > 350 mg/24 hour; subsequently, all their manifestations and the available follow-up after treatment were recorded.
The pathology of kidney biopsy of these patients was re-evaluated by the pathologist involved in this study.
Bioethical standards
Ethical approval and consent to participate: the study with project number of 24413 in Shiraz University of Medical Sciences was approved by the Human Ethics Review Committee of our University of Medical Science with the code number: IR.sums.med.rec.1400.525.
Inclusion criteria
The study group members were enrolled based on New 2017 EULAR/ACR Classification Criteria for adult and juvenile IM, which sub-classified the patients into 4 main sub-groups including DM, ADM, JDM, and PM using the classification tree.
It divided the patients into definite (probability cut-off ≥ 90%), probable (probability ≥ 55%), possible (probability ≥ 50%, but < 55%), and improbable/disposable (probability < 50%), depending on the score obtained by adding the selected variables (http://www.imm.ki.se/biostatistics/calculators/iim).
We included the definite IIM (probability ≥ 90) in the sub-group of DM patients with disease duration of ≤ 12 months [3]. All included patients required available renal function tests including BUN and creatinine and urine analysis.
The patients with positive protein in their urine analysis were checked for 24-hour urine protein and creatinine, and patients with the presence of proteinuria > 350 mg/24 hours were included and their biopsies were reviewed by the pathologist involved in this study. The patients were also followed for their treatment response.
Exclusion criteria
The patients with other causes of myopathy including PM, JIM, IBM, and IMNM, and those who had manifestations of overlap myositis with signs and symptoms or specific tests of other connective tissue diseases like SLE, systemic sclerosis (SSc), rheumatoid arthritis (RA), mixed connective tissue disease (MCTD), or Sjögren’s syndrome (SS), and the patients with cancer-associated IIMs, amyopathic DM (ADM), and antisynthetase syndrome (AS) were excluded as well.
All clinical manifestations of the patients and their laboratory data were recorded. Lab test results, capillaroscopy parameters based on the European League Against Rheumatism/Scleroderma clinical trials consortium consensus [8], kidney biopsy reports, and follow-up of these patients for treatment response were also evaluated.
Results
In our study, the profiles of 205 referred patients were evaluated (Fig. 1). There were 74 patients who fulfilled the definite criteria for DM of ≤ 12 months’ duration. Among the excluded patients, we also evaluated their kidney profiles, and there was no case of proteinuria in all groups including the ADM, AS, hereditary, overlap, JDM, PM, DM > 12 months, SLE, MCTD, or statin-induced groups. There was only one patient with chronic kidney disease secondary to ureteral stenosis (post bladder carcinoma surgery) one year prior to DM symptoms.
Fig. 1
Diagram of studied patients with myopathy: the total number at the study beginning and the excluded ones.
The dermatomyositis patients comprised 52 females and 22 males, with a median age of 37 (19–65) years and a median disease duration of 4.5 (1–12) months. All of them had active disease including active skin manifestations of DM along with muscle weakness and/or myopathy in EMG study. Twenty-six (35%) patients had lung involvement at the time of the study.
Extractable nuclear antigen studies were available for 25 patients, and only 5 patients had positive results. Anti-Ro was positive in these 5 patients and anti-LA in one patient along with anti-Ro positivity. About 88% of patients with DM were not on any treatment before being included in this study.
Only 9 patients (12%) were on medication. From them, 6 patients were on prednisolone, 2 on azathioprine, 2 on methotrexate, and one patient was on cyclosporine treatment. One patient had diabetes mellitus and was on metformin therapy, and 2 patients, due to hyperlipidaemia, were on statin treatment. Only 2 (2.7%) patients with DM had proteinuria and bland urine sediment on their arrival during acute onset of the disease.
There were no cases of AKI or chronic kidney damage based on the KDIGO guideline and no case of rhabdomyolysis. If we included the patients with PM, ADM, AS, DM more than 12 months, and JDM in our study, again the incidence of proteinuria was very low: 2/160 (1.25%).
Case 1
A 25-year-old man referred with a 2-month history of puffy fingers and scaly skin lesions of the knuckles of both hands, especially his metacarpophalangeal joints (MCP), and then after 1 month with bilateral upper and lower extremities proximal muscle weakness, dysphagia, and nasal speech. The patient mentioned 14-kg weight loss during this time. He had no history of Raynaud’s phenomenon or dyspnoea at that time.
On examination, he had significant periorbital oedema, multiple Gottron’s signs on both MCP joints and knuckles, both lower extremities 2+ pitting oedema, and significant proximal muscle weakness. The laboratory tests are shown in Table I.
Table I
Clinical, laboratory, capillaroscopy, and medication details of the first patient with dermatomyositis and proteinuria
[i] ALK – anaplastic lymphoma kinase, ALT – alanine transaminase, ANA – anti-nuclear antibody, AST – aspartate aminotransferase, BUN – blood urea nitrogen, CPK – creatine phosphokinase, CRP – C-reactive protein, CT – computed tomography, dsDNA – double-stranded DNA, EMG–NCV – electromyography (EMG) and nerve conduction velocity (NCV), ENA – extractable nuclear antigens, ESR – erythrocyte sedimentation rate, Hb – haemoglobin, LDH – lactate dehydrogenase, serum Alb – serum albumin, WBC – white blood cells.
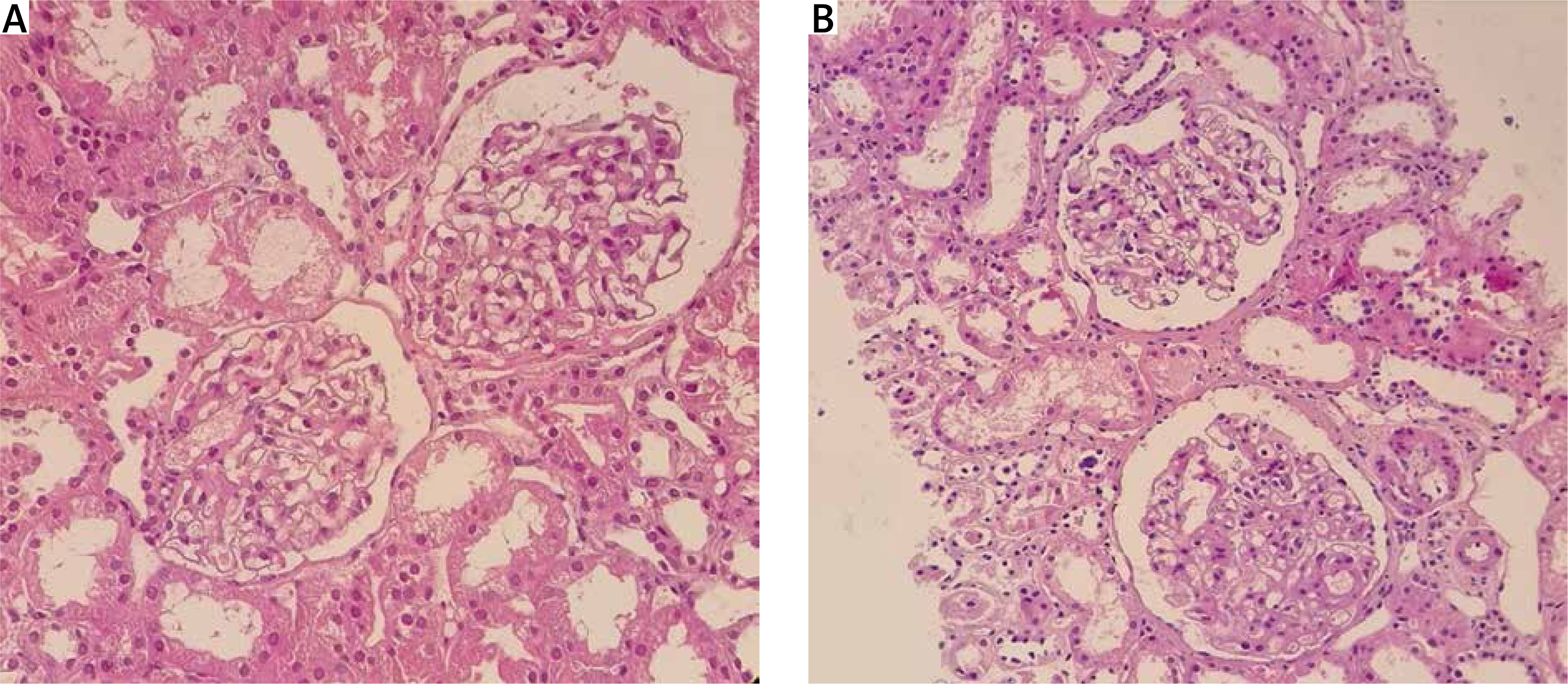
A new diagnosis of hypothyroid was also made with negative anti-thyroid peroxidase (anti-TPO) antibody. Due to significant proteinuria and hypoalbuminaemia, a kidney biopsy was performed, which showed mild focal mesangial cell hypercellularity and expansion with no immune deposit in immunofluorescence study in favour of focal mesangioproliferative GN (mesPGN; Fig. 2). Treatment was started using methylprednisolone pulse therapy 1 g for 3 days, intravenous immunoglobulin (IVIG) monthly, mycophenolate mofetil 1500 mg daily, tacrolimus 2 mg daily, and levothyroxine and losartan.
Fig. 2
Kidney biopsies from case 1 (A) and case 2 (B), showing glomerulus with mild mesangial hypercellularity and mild increase in the mesangial matrix (light microscopy, haematoxylin and eosin, 400 ×).
After 5 months of follow-up the patient still had high creatine phosphokinase (CPK): 2715 and lactate dehydrogenase (LDH): 1143 on tapering of prednisolone to 7.5 mg. Therefore, rituximab (RTX) was started with 2 × 1 g during 2 weeks.
After 6 months, patient’s muscles weakness significantly improved, and muscle enzymes became normal. Rituximab continued for another dose, and then it was stopped and the medication was continued with azathioprine 100 mg per day, tacrolimus 1 mg daily and prednisolone 5 mg along with levothyroxine.
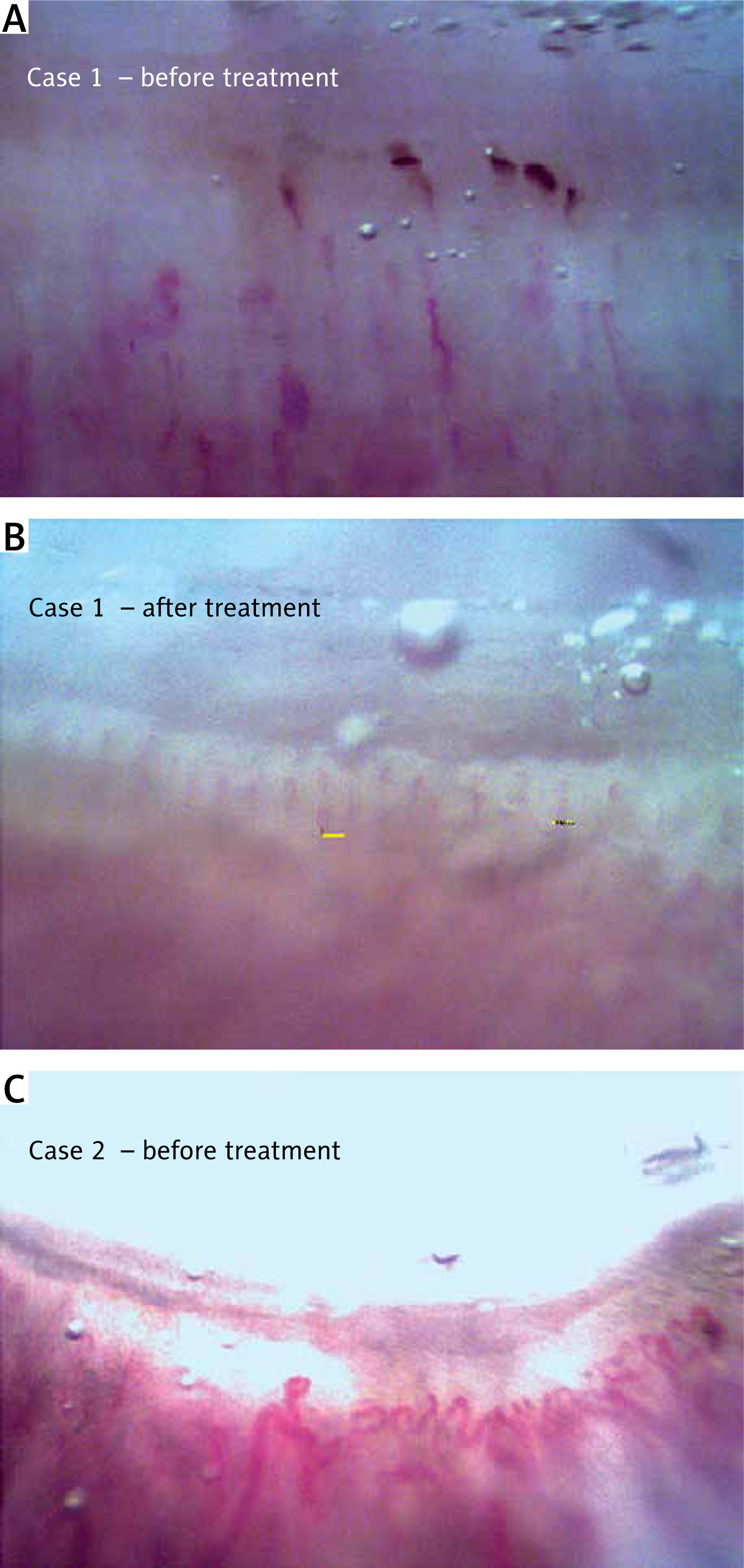
After nearly 3 years, the patient had normal muscle power, very mild remaining nasal speech, and no dysphagia or proteinuria were observed. Also capillaroscopy image showed improvement (Fig. 3, Table I).
Case 2
A 28-year-old man referred with a 3-month history of multiple skin lesions of extremities and facial oedema, and then gradually during one month developed proximal muscle weakness, myalgia, and dysphagia. Patient had a history of mild cough and dyspnoea; COVID-19 PCR test was performed 3 times, and all tests were negative.
On examination, the patient had periorbital oedema, heliotrope rash, holster and Gottron’s signs, and significant proximal muscle weakness. All muscles were tender and swollen; the patient had polyarthritis of all proximal interphalangeal, wrist, and ankle joints. In patient’s work-up study, bilateral lung infiltration resembling COVID-19 lung involvement including multiple ground glass appearance (GGO), fibrosis, and significant proteinuria were seen. In naifold capillaroscopy active scleroderma pattern was found (Fig. 3).
Kidney biopsy showed mild mesangial hypercellularity and a mild increase in mesangial matrix with trace deposition of IgG and IgM immune complex in the mesangium in favour of focal mesPGN (Fig. 3). Mycophenolate mofetil, methylprednisolone, and tacrolimus were started along with monthly IVIG.
After 4 months, proteinuria and muscle power significantly improved. However, after 8 months the patient failed to attend his follow-up and decreased mycophenolate mofetil to 500 mg daily and did not use tacrolimus. The patient developed recurrence of disease with ulcerative Gottron papules, heliotrope rush, increasing weakness, proteinuria, and dysphagia along with pneumomediastinum, so treatment was restarted including cyclophosphamide, IVIG, and methylprednisolone (Table II).
Table II
Clinical, laboratory, capillaroscopy, and medication details of the second patient with dermatomyositis and proteinuria
[i] ACPA – anti-cyclic citrullinated peptide antibody, ALK – anaplastic lymphoma kinase, ALT – alanine transaminase, ANA – anti-nuclear antibody, AST – aspartate aminotransferase, BUN – blood urea nitrogen, CPK – creatine phosphokinase, CRP – C-reactive protein, CT – computed tomography, dsDNA – double-stranded DNA, EMG–NCV – electromyography (EMG) and nerve conduction velocity (NCV), ENA – extractable nuclear antigens, ESR – erythrocyte sedimentation rate, Hb – haemoglobin, LDH – lactate dehydrogenase, RF – rheumatoid factor, WBC – white blood cells.
Discussion
We evaluated the profile of patients with early (first year) active DM to find the kidney manifestations of these patients, followed them, and found a low prevalence of GN in these patients with good prognosis after treatment.
Dermatomyositis is a kind of IIM in which kidney involvement, like other connective tissue diseases (e.g. SLE or MCTD), is not a usual clinical manifestation, and kidney involvement is mostly reported as a case series.
In a study in Japan that was carried out on 20,523 registered kidney biopsy cases, after excluding patients with SLE, a total of 110 subjects (0.5%) with connective tissue diseases were selected. There was only one patient with PM/DM who showed endocapillary proliferative GN with IgA deposition [5].
Two retrospective series of patients with IIMs reported a prevalence of renal disease between 21 and 23% in patients with DM/PM. Two types of kidney involvement were the most common. The first form was acute renal failure that might have been caused by rhabdomyolysis, drug toxicity, or infection; the less frequent form was an association with chronic GN [7–9].
The first study, which was done in 2005 on 65 Taiwanese patients, with a ‘definite’ diagnosis of PM/DM showed that 14 (21.5%) (5 PM and 9 DM) patients had variable degrees of renal involvement. All patients had varying degrees of haematuria and proteinuria. Five out of the 9 DM and 4 out of 5 PM patients developed acute tubular necrosis with renal failure related to acute rhabdomyolysis and needed emergent haemodialysis. The other patients mostly had chronic GN in their biopsies.
In the PM group, only 1 out of 5 patients had moderate proteinuria that also showed an overlap of SLE. In the DM group, 2 out of 9 patients showed moderate to heavy proteinuria, one of whom had an overlap with SLE and the other one showed IgA nephropathy in kidney biopsy. There was high mortality in the rhabdomyolysis/ATN group in the follow-up, and some of them were cancer-associated DM/PM or hepatitis-associated [10].
In another retrospective study [9] that was done on 150 patients diagnosed with DM (96; 64%), PM (26; 17%), or AS (28; 19%), renal involvement occurred in 35 (23.3%) patients: 21 patients (22%) with DM, 6 (23%) with PM, and 8 (29%) with AS. Acute kidney injury was observed in 16 (10.7%) patients and CKD in 31 (20.7%) patients. Proteinuria was > 0.3 g/day in 26/86 patients (30%).
Also, AKI was seen mostly in the first 6–12 months after the diagnosis and was mostly related to drug toxicity, rhabdomyolysis/ATN associated with severe acute heart failure, and one thrombotic microangiopathy. No significant association was found between the type, severity, and level of CPK and the occurrence of AKI. Advanced age, diabetes, and cardiac disease were associated mostly with AKI.
Chronic kidney disease (CKD) was noted in 31 (20.7%) patients, and higher age at IIM onset, male gender, history of cardiovascular events, and a previous occurrence of AKI were identified as the risks factors of CKD.
Fourteen patients (6 PM, 5 DM, and 3 AS) underwent kidney biopsy in the presence of proteinuria (n = 13), and/or AKI (n = 5), and/or CKD (n = 3). There were 4 patients with DM (n = 3) and AS (n = 1) in the first 6 months of the disease, and 2 DM patients showed minimal change disease (MCD); one of them received cyclosporin and azathioprine and developed CKD, and the second received methotrexate and IVIG and went into remission.
One patient with DM had vascular lesions that led to CKD in the follow-up. One AS patient had IgA nephropathy and received steroid, methotrexate, IVIG, and tumor necrosis factor inhibitors and went into remission.
They also gathered the previous reports on patients with early DM and AS in their 6 months of disease and found 5 patients (3 DM and 2 AS), 3 of whom showed IgA nephropathy, one with pauci-immune crescentic glomerulonephritis, and one with membranoproliferative GN, including the first study mentioned previously [9].
In a study done on 107 patients with DM/PM, 26 (24.3%; 22 patients with DM and 4 with PM) were found to have suffered varying degrees of renal involvement. Acute kidney injury was seen in 8 (7.48%) and CKD in 18 (16.82%) patients. The kidney biopsies of 16 patients showed chronic tubulointerstitial nephritis (7 cases), minimal change disease (2 cases), focal segmental glomerulosclerosis lesions (3 cases), IgA nephropathy (2 cases), membranous nephropathy (1 case), and amyloid deposits (1 case). They showed that these manifestations resulted from drug and myoglobin-induced renal damage as well [11].
Another point in favour of several pathogenic renal involvement processes is that the reported immunofluorescences (IF) are highly varied. In some cases of membranous nephropathy the IF for IgG and C3 was positive, as in the well-known idiopathic form, and in the other cases full positivity for IgA, IgG, IgM, C1q, and C3 was seen. The same has been observed in mesangial proliferative GN, which has been categorized as IgA nephropathy in some instances due to the dominance of IgA at immunofluorescence, while other patients showed a broad range of immunoglobulins and complement deposition [6].
In our study, we found low prevalence (2.7%) of GN in the type of mesangioproliferation, with only one patient having a positive IF study from the patients with active DM with the duration of less than one year. According to research [6], GN in IIM patients was typically caused by immune complex deposition in the glomeruli.
In individuals with IIMs, who underwent renal biopsy, a wide range of immune-complex glomerulonephritis has been documented [6, 7]. As with muscle biopsies, DM and PM patients are likely to have diverse and specific images at renal biopsy, indicating the activation of either humoral or cell-mediated immunity.
In this regard, it has been suggested by Cucchiari et al. [6] that PM patients have a picture of mesangial proliferative GN, whereas DM patients have membranous nephropathy more frequently. This, however, is not always the case. There have been reports of PM patients developing membranous nephropathy and DM patients developing mesangial proliferative GN, as in our cases.
Although mesangial proliferative GN can clinically present with haematuria (even gross haematuria), the most common clinical presentation of this glomerulopathy is proteinuria, and nephrotic syndrome and diffuse foot process fusion are usually demonstrated in electron microscopy, suggesting that this disorder may be part of the minimal change disease (MCD-FSGS) spectrum. Although the renal prognosis is good for patients with isolated haematuria, it is not as good for those with nephrotic syndrome because 10–30% may develop progressive renal insufficiency.
In a retrospective cohort study of 140 patients with biopsy-proven mesangial proliferative GN, renal survival was negatively associated with age at baseline, serum creatinine, the amount of proteinuria, hypoalbuminaemia, high blood pressure, and male gender [12].
As in our cases, both patients presented with significant proteinuria, hypoalbuminaemia, and bland urine sediment with normal kidney function; in the follow-up, our first patient’s proteinuria responded after 6 months of follow-up to prednisolone, mycophenolate mofetil, tacrolimus, and IVIG treatment although his muscular manifestations required rituximab treatment.
In the 3-year follow-up, the patient had no active disease or kidney involvement. His capillaroscopy also became normal and muscle power significantly improved, but mild nasal speech remained. The second patient, after 3 months, showed a good response to prednisolone, mycophenolate mofetil, tacrolimus, and IVIG treatment with complete recovery of his kidney and muscle manifestations. However, due to losing his regular follow-up and discontinuation of his main drugs the patient had recurrence of all symptoms and treatment was restarted.
In their active phase of the disease, both patients had skin manifestations, muscle weakness, abnormal electromyography, high ESR and CRP, and high anti-Ro positivity. Both had abnormal shaped capillaries, dilated loops, and haemorrhage in their capillaroscopy; both patients had good prognosis and response of their kidney function, muscle weakness, and laboratory tests after full treatment in their follow up. One patient had capillaroscopy follow-up, and all abnormalities were resolved in all 8 fingers under nailfold capillaroscopy.
Therefore, we found a low prevalence of GN in the pattern of mesangioproliferative GN in our DM patients in the early active phase of the disease, without any other cause or drug before being in this study. Also, in our excluded PM, ADM, AS, DM more than 12 months, and JDM groups, we did not find any GN.
Study limitations
The limitation of the study was that only patients with acute and active phase of the disease mostly without receiving any previous medications were included. Also, they had a low number of cardiovascular risk factors, so maybe it was the result of a lack of AKI or CRF in this study; further studies in their follow-up and inclusion of higher numbers of patients would be helpful.
Conclusions
A rare extra-muscular manifestation of active dermatomyositis is GN and proteinuria mostly in the form of mesangioproliferative GN. This presentation of dermatomyositis is usually responsive to aggressive immunosuppressive treatments and has good early prognosis.
Although we found that kidney involvement was rare in patients with DM, it can be a manifestation of the active phase of the disease in the mesangioproliferative type of GN and should be considered as a manifestation also of the inflammatory phase of this disease. Full immunosuppressive treatment showed early recovery in these patients.