
Contrary to the common trend in the world of medicine, where specializations are narrowing down not only to particular systems or organs, but even to specific diseases, internal medicine and rheumatology remain a thoroughly interdisciplinary fields. Recently, a relatively new entity – the VEXAS (vacuoles, E1 enzyme, X-linked, auto-inflammatory, somatic) syndrome, due to its clinical manifestations and associations with already long-known diseases (Table I), particularly draws the interest of internists, rheumatologists and hematologists [1, 2].
Table I
The main VEXAS syndrome associations with other already known/well-defined diseases
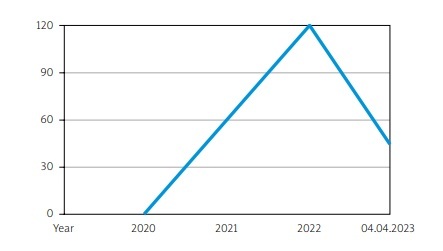
The growing interest in VEXAS syndrome can be seen in the number of published works on this subject over the last 3 years (Fig. 1).

Thanks to the discovery of VEXAS syndrome, the connection of somatic mutations with clonal hematopoiesis, which causes a systemic inflammatory response, seems more obvious. We are getting closer to the explanation of the etiopathogenesis of autoinflammatory diseases, but also faced the possibility of separating them into a new group, known in the literature as hematoinflammatory diseases [3].
What exactly is VEXAS syndrome? The term VEXAS is an acronym for a monogenic auto-inflammatory disease caused by somatic mutations located on the X chromosome in the UBA1 (ubiquitin like modifier activating enzyme 1) gene, which encodes E1 enzyme, a protein involved in ubiquitin activation. A characteristic feature observed in bone marrow biopsies is the presence of vacuoles in the cytoplasm of myeloid and erythroid progenitor cells [1]. The dysfunction of the ubiquitination process results in a systemic inflammatory response manifested by fever, weight loss, and a variety of local and systemic symptoms in which several specialists are often involved before having a suspected diagnosis or the genetic confirmation of VEXAS syndrome.
Is VEXAS syndrome really infrequent? Above all, despite low awareness among physicians and the seemingly anecdotal prevalence of the disease, it still should be suspected and considered a potential diagnosis! However, according to an analysis published in early 2023 in the Journal of the American Medical Association (JAMA), the disease affects 13,200 men and 2,300 women over 50 years of age, which means 1 in 4,269 American men and 1 in 26,238 American women in this age group [4]. For comparison, the incidence of granulomatosis with polyangiitis (GPA) in the United States is lower than for VEXAS syndrome, since it is estimated to be around 9.9 cases per million person-years [5].
Therefore, who should be suspected of having VEXAS syndrome? The average patient with VEXAS syndrome is a man (due to the protective role of the unmutated allele in women), over 50 years old (the age at which the somatic mutation is presumably produced), with remarkable hematologic disorders, of which macrocytic anemia is the most constantly observed, and a widespread spectrum of single or combined inflammatory symptoms, for which patients have previously been diagnosed with auto-immune diseases that include relapsing polychondritis, neutrophilic dermatoses and different types of systemic vasculitis involving various vessel sizes. All these disorders are usually refractory to small-to-medium doses of glucocorticosteroids and different conventional and biologic immunosuppressive agents.
Ferrada et al. [6] proposed a diagnostic algorithm for patients with a previous diagnosis of relapsing polychondritis based on identifying men with a mean red blood cell volume above 100 fl and a platelet count below 200,000/μl, allowing an effective diagnosis of VEXAS syndrome with a sensitivity of 100% and a specificity of 96%.
When should the genetic study be performed? Genetic testing (searching for UBA1 gene mutations) is recommended in patients with refractory cytopenias and other inflammatory and autoimmune manifestations of unknown etiology. However the results should be interpreted only when analyzing the entire clinical picture. As much as a final genetic test may pose a challenge in daily clinical practice, treatment of VEXAS syndrome seems to be even more challenging and demanding.
Which drugs have proven efficacy? High-dose glucocorticosteroid therapy continues to play a crucial role and the prognosis is still unfavorable, with a 5-year survival rate of approximately 63% [7]. Georgin-Lavialle et al. [7] found in their retrospective study that factors associated with a poor prognosis include involvement of the digestive system, pulmonary infiltrates and enlargement of mediastinal lymph nodes. As potential therapeutic armamentarium, conventional immunosuppressive drugs (methotrexate, azathioprine, mycophenolate mofetil or cyclophosphamide) as well as monoclonal antibodies targeting interleukin-6 (IL-6), IL-1 or tumor necrosis factor α (TNF-α), B-cell depleting agents (e.g. rituximab) and Janus kinase (JAK) inhibitors have been reported to be associated with partial or no responses [8]. However, ruxolitinib, a JAK1/2 inhibitor, has recently demonstrated favorable clinical and glucocorticosteroids-sparing effects in a retrospective study [9].
Among hematologic treatments, azacitidine, an hypo-methylating agent has shown good results, particularly in patients with VEXAS syndrome and myelodysplastic syndromes. Finally, allogeneic hematopoietic stem-cell transplantation is considered the only potential curative therapy in VEXAS syndrome nowadays [8].
Randomized clinical trials with different agents, including proinflammatory cytokine blockers, JAK inhibitors and azacitidine are still needed to better determine the real response rate and the phenotypes in which they could be particularly useful. In adiition, in order to identify which patients will benefit from bone marrow transplantation, a phase II clinical trial studying the safety of allogeneic hematopoietic stem-cell transplantation in patients with VEXAS syndrome is ongoing (ClinicalTrials.gov identifier: NCT05027945).
Do we need collaborative networks? Because the clinical spectrum of VEXAS syndrome is still growing and the therapeutic options are really scarce, there is a need to collect more information on patients with VEXAS syndrome to better understand the cause and consequences of this disease, as well as the most useful therapeutic strategies. In this sense, the Autoinflammatory Diseases Alliance (AIDA) Network is a multicenter and international registry created to gather all the information on VEXAS syndrome and other autoinflammatory conditions [10].
Should we look ahead in the field of autoinflammatory diseases in adult patients? In the future, VEXAS syndrome along with other related autoinflammatory conditions, and maybe other hematoinflammatory diseases, in adulthood may change our perception of the already supposedly known rheumatic or systemic autoimmune diseases, and genetic diagnosis may become a routine clinical practice for physicians in experienced referral centers.
Conclusions
In 2020 VEXAS, a new autoinflammatory syndrome was defined.
Men older than 50 years are more susceptible to VEXAS syndrome than women.
Diagnosis of VEXAS syndrome is based on clinical and biological (laboratory and hematologic) manifestations and confirmation of postzygotic mutations in the UBA1 gene.
This syndrome appears to be much more common than initially assumed.
Although most patients respond to high-dose glucocorticosteroids, most conventional and biologic immunosuppressive agents fail to control the disease activity and progression.
Azacitidine, a hypomethylating agent, has shown effectiveness, mainly in patients with myelodysplastic syndromes.
To date, allogeneic hematopoietic stem-cell transplantation is considered the only potential curative therapy in VEXAS syndrome.