
Introduction
Calcium is a fundamental constituent of the human body and has both intracellular and extracellular functions. Within the cell, calcium contributes to the homeostasis of many processes, including growth, proliferation and apoptosis [1].
Outside the cell, calcium represents an essential component of the mineral matrix of bone, contributes to blood coagulation and is required for the correct functioning of the neuronal and muscular apparatus [2].
The concentration of extracellular calcium is maintained within a strict interval and variations are promptly corrected by the intervention of hormones. Similarly, the intracellular concentration of calcium is constantly kept at a low level, and stimuli inducing a peak give rise to several cascades, culminating in the activation of enzymes and transcriptional pathways.
Acting as a second messenger, calcium plays a central role in inflammation, and its aberrant deposition in extracellular matrix (ECM), occurring during dystrophic or reparative processes, is at the basis of ectopic calcifications in either skeletal or extra-skeletal sites.
Ankylosing spondylitis (AS) is a rheumatic disease characterized by inflammation and the final calcification of the entheses, which are the site of bone attachment of tendons, joint capsules and ligaments [3]. Ankylosing spondylitis belongs to the group of spondyloarthropathies (SpA), with which it shares a common genetic signature, pathogenesis, clinical manifestations and therapeutic management [4, 5].
The most relevant features of SpA are the predominant involvement of the axial skeleton, including spine, pelvis and rib cage (although peripheral joints, especially of the lower limbs, can be asymmetrically involved), a strong genetic predisposition partly related to the presence of the genetic marker HLA-B27, the absence of the rheumatoid factor, the occurrence of enthesitis and dactylitis, the evidence of bone apposition rather than bone erosion on X-rays, and a corollary of associated extra-articular manifestations, including uveitis, psoriasis or inflammatory bowel diseases (IBD).
The pathogenesis of SpA is multifactorial. According to one of the most accredited theories, HLA-B27 heavy chain misfolding would eventually induce endoplasmic reticulum stress, interfere with antigen presentation, and aberrantly activate natural killer (NK) cells, CD8+ and CD4+ T lymphocytes. This process may be amplified by episodes of disturbance of the intestinal microbiome (dysbiosis) [4] or mechanical stress [3], generating pro-inflammatory cascades unleashed by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular pattern (DAMPs).
Activated macrophages and dendritic cells would promote the differentiation of Th17 lymphocytes and the final secretion of cytokines such as interleukin (IL)-17, IL-22, IL-23 and tumor necrosis factor-alpha (TNF-α), in turn involved in bone remodeling [4]. These immunologic pathways have been exploited as pharmacological targets for the formulation of biologic drugs, such as anti-TNF and anti-IL-17 agents, whose efficacy has been shown in both axial and peripheral forms of SpA, as well as in SpA-associated extra-articular manifestations [5, 6].
Ankylosing spondylitis, whose prevalence is 0.1–0.5% in the Caucasian population [6], usually develops in HLA-B27 positive young males (female/male ratio 1/9) and mainly affects the spine and sacroiliac joints.
The most typical symptoms include protracted morning stiffness, inflammatory low-back pain and spine morphofunctional changes, leading to progressive disability. The classical course of the disease is characterized by a slow progression from an early non-radiographic axial spondyloarthritis stage (nr-axSpA), in which entheseal and bone inflammatory signs are solely detectable by magnetic resonance imaging (MRI), to a late radiographic stage, in which soft tissue ossification (e.g. syndesmophyte formation and spine fusion) can be revealed by conventional X-rays.
Those forms of axial SpA not developing skeletal structural changes are classified as nr-axSpA. Though ~12% of nr-axSpA subjects can develop AS in the first two years of the disease, it is unclear whether nr-axSpA might be considered as an early phase of AS or rather represents a separate clinical entity [7].
Ankylosing spondylitis is characterized by a bone paradox mirrored, on the one hand, by the development of trabecular and cortical osteoporosis and, on the other hand, by the overproduction of cortical bone and entheseal ossification [8].
Reiterated micro-traumatisms and an inflammatory insult of enthesis, whose main function is to redistribute mechanical stress [3], have been hypothesized to be at the basis of enthesitis and calcification. Notably, entheseal calcification is not specific to SpA, being also found in other rheumatic, metabolic and post-traumatic conditions, including diffuse idiopathic skeletal hyperostosis (DISH) and crystal-associated enthesopathy [9].
Despite the tendency to bone accretion, the risk of osteoporosis in AS is increased, presumably on the basis of chronic inflammation, hypo-mobility and malabsorption, thus making calcium and vitamin D supplementation often necessary [10].
However, the long-term benefits of calcium supplementation are still unclear. It may be hypothesized that an unbalanced calcium metabolism may worsen the process of calcification at the entheses, especially if other mineral defects (such as an altered phosphate homeostasis) [2] or a deficit in ECM repair coexist.
Since there is still scarce evidence on the usefulness of calcium supplementation in these patients and international guidelines are lacking, we aimed to provide a narrative review of the current literature focusing on calcium absorption, metabolism, function and bone homeostasis in AS patients compared to healthy subjects. Furthermore, papers concerning the effects of calcium supplementation in AS were also searched and discussed.
We selected 65 English-written papers dated from 1979 to 2020, by means of a search of the PubMed database, using the combination of words: “calcium”, “vitamin D”, “mineral metabolism”, “ankylosing spondylitis”, “enthesitis”, “osteoporosis”. Results are reported in the next paragraphs and summarized in Table I.
Table I
Calcium metabolism in ankylosing spondylitis patients and in healthy subjects – main studies of calcium supplementation in ankylosing spondylitis
| Calcium metabolism | Healthy subjects | Ankylosing spondylitis patients | Main results of preclinical or clinical studies on calcium metabolism in ankylosing spondylitis | In favor of calcium supplementation in ankylosing spondylitis patients | |
|---|---|---|---|---|---|
| Preclinical models and human laboratory studies | Clinical studies | ||||
| Extracellular calcium concentration | Within the range 1.1–1.3 mmol/l, ionized form | Usually within the range 1.1–1.3 mmol/l, ionized form | – | Normal corrected calcemia in AS and RA patients and hypercalcemia in subjects with polymyalgia rheumatica (Bontoux et al. [25] 1979; case control study) No significant differences in serum calcium levels between AS patients and controls (Cai et al. [26] 2015; meta-analysis) Higher serum calcium levels in AS patients with urolithiasis compared to those without (Gönüllü et al. [58] 2017; prospective case control study) | No |
| Intracellular calcium concentration | Within the normal range (100 nM) and tightly controlled | Increased in immune cells where it presides over the activation of pro-inflammatory pathways | Enhanced intracellular calcium entrance in neutrophils of AS patients, prevented by treatment with infliximab (Ugan et al. [30] 2016) Anti-inflammatory properties of blockade of IKCa1 lymphocyte potassium channels, which regulate calcium influx in CD4+ and CD8+ T cells of AS patients (Toldi et al. [31] 2016) Loss of physiological intracellular calcium oscillations in peripheral monocytes of patients with SpA (especially AS) compared to controls; possible association with differentiation towards activated phenotypes (Moz et al. [29] 2020) | – | No |
| Calcium intestinal absorption | 15–30% of the dietary intake, mediated by either active or passive absorption | Reduced in patients with inflammatory bowel diseases, dysbiosis and gut subclinical inflammation | Reduced colonic expression of the L-type calcium channel Cav1.3 in trinitrobenzene sulfonic acid colitis mice; IFN-γ-induced transcriptional and posttranscriptional repression of the Cav1.3 gene in HCT116 cells (Radhakrishnan et al. [19] 2016) Downregulation in duodenal epithelial cells of TNFΔARE mice of the calcium transporters TRPV6, calbindin D9K and PMCA1b, associated with increased trabecular and cortical bone resorption (Huybers et al. [20] 2008) Reduction in colonic inflammation, intestinal permeability and diarrhea in HLA-B27 transgenic rats supplemented with high calcium doses for 7 weeks; reduced expression of mucosal IL-1β and increased expression of ECM remodeling genes in calcium-fed rats compared to controls (Schepens et al. [52] 2010) | Lower calcium intake in IBD patients resulting from the 22-item quantitative validated frequency food questionnaire compared to controls; association with self-reported lactose intolerance (Vernia et al. [23] 2014; case control study) Higher calcium intake in non-IBD AS patients compared to controls measured through the semi-quantitative food frequency questionnaire (Soleimanifar et al. [24] 2018; case control study) | Yes |
| Calcium resorption from bone | Related to physiologic bone remodeling (about 20% of total bone mass) | Increased in response to pro-inflammatory cytokine secretion | Increased trabecular and cortical bone resorption observed on tomography scanning in TNFΔARE mice, presumably compensating calcium malabsorption or loss in the urinary tract; down-regulation of calbindin-D9K mRNA in bone (Huybers et al. [20] 2008) | Low BMD detected in 13% out of 332 recruited early SpA patients; significant association with inflammatory lesions in MRI, ESR and CRP values and male gender (Briot et al. [47] 2013; longitudinal prospective cohort study) Reduced spinal BMD in patients with nr-axSpA compared to subjects with mechanical low back pain; positive association between low spinal and femoral BMD and lumbar inflammatory findings in MRI in nr-axSpA individuals (Akgöl et al. [46] 2014; case control study) Lower cortical vBMD in radius and lower trabecular vBMD in tibia in AS patients compared to healthy controls detected on HR-pQCT; vertebral fractures associated with lower lumbar cortical vBMD and thinner cortical bone in radius and tibia; inverse correlation between mSASSS and trabecular vBMD in lumbar spine radius and tibia (Klingberg et al. [48] 2013; case control study) Reduced radius cortical vBMD in early nr-axSpA patients versus controls detected through HR-pQCT; lower trabecular vBMD in female gender (Neumann et al. [49] 2018; case control study) | Yes |
| Calcium resorption from kidney | About 90% of calcium filtered | Unknown (kidney stones related to hypercalciuria?) | Down-regulation of the calcium transporters calbindin-D 28K and NCX1 in kidney epithelial cells of TNFΔARE mice, paralleled by increased trabecular and cortical bone resorption (Huybers et al. [20] 2008) | No difference in urinary calcium excretion between AS patients with and without nephrolithiasis (Rezvani et al. [56] 2018; retrospective cohort study) Renal stones diagnosed in 25% of AS patients versus 3.3% of healthy controls; positive association between the risk of renal stones and patients’ age, disease duration and hypercalciuria (Korkmaz et al. [57] 2005; case control study) No significant association between high urinary calcium excretion and risk of urolithiasis in AS patients (Gönüllü et al. [58] 2017; prospective case control study) | No |
| Serum PTH levels | Within the range 10–60 pg/ml | Normal or higher | – | No significant differences in serum PTH levels between AS patients and controls (Cai et al. [26] 2015; meta-analysis) No significant differences in serum PTH levels between AS patients and controls; no association with the risk of urolithiasis (Gönüllü et al. [58] 2017; prospective case control study) Higher serum PTH levels in AS patients compared to controls; positive correlation with serum levels of DKK-1 (Orsolini et al. [43] 2018; case control study) Inverse correlation between serum PTH levels and AS disease activity, measured through ESR and CRP values and BASDAI scores (Lange et al. [27] 2001; case control study) Normal serum values of PTH in nr-axSpA patients compared to controls (Akgöl et al. [46] 2014; case control study) | Yes |
| Serum 25(OH) vitamin D levels | Within the range 30–60 ng/ml | Reduced | – | Reduced serum level of 25(OH) vitamin D in AS patients compared to controls; significant association between lower 25(OH) vitamin D levels and higher ESR values (Cai et al. [26] 2015; meta-analysis) Normal values of serum 25(OH) vitamin D in nr-axSpA patients (Akgöl et al. [46] 2014; case control study) | Yes |
| Serum 1,25(OH)2 vitamin D levels | Within the range 25–45 pg/ml | Within the range 25–45 pg/ml | – | Normal serum concentration of 1,25(OH)2 vitamin D in AS patients; inverse association with disease activity evaluated through ESR, serum CRP values and BASDAI scores (Cai et al. [26] 2015; meta-analysis) Negative association between serum 1,25(OH)2 vitamin D levels and AS disease activity, measured through ESR and CRP values and BASDAI scores; positive correlation with bone alkaline phosphatase (Lange et al. [27] 2001; case control study) | No |
[i] AS – ankylosing spondylitis, BASDAI – Bath ankylosing spondylitis disease activity index, BMD – bone mineral density, vBMD – volumetric BMD, CRP – C-reactive protein, ECM – extracellular matrix, ESR – erythrocyte sedimentation rate, HLA-B27 – human leukocyte antigen-B27, HRpQCT – high-resolution peripheral quantitative computed tomography, IBD – inflammatory bowel disease, IFN-γ – interferon-gamma, IL – interleukin, NCX1 – Na+/Ca2+ exchanger, PMCA1b – plasma membrane calcium-transporting ATPase 1b, PTH – parathyroid hormone, RA – rheumatoid arthritis, mSASSS – modified Stoke Ankylosing Spondylitis Spine Score, SpA – spondyloarthritis, nr-axSpA – non-radiographic axial spondyloarthritis, TNF – tumor necrosis factor, TRPV – transient receptors potential vanilloid calcium channel.
Physiological calcium metabolism
Calcium is an electrolyte governing a wide range of extracellular and intracellular functions, including blood coagulation, nerve functioning, muscle contraction, bone scaffolding, exocytosis, cell proliferation, motility and apoptosis [2].
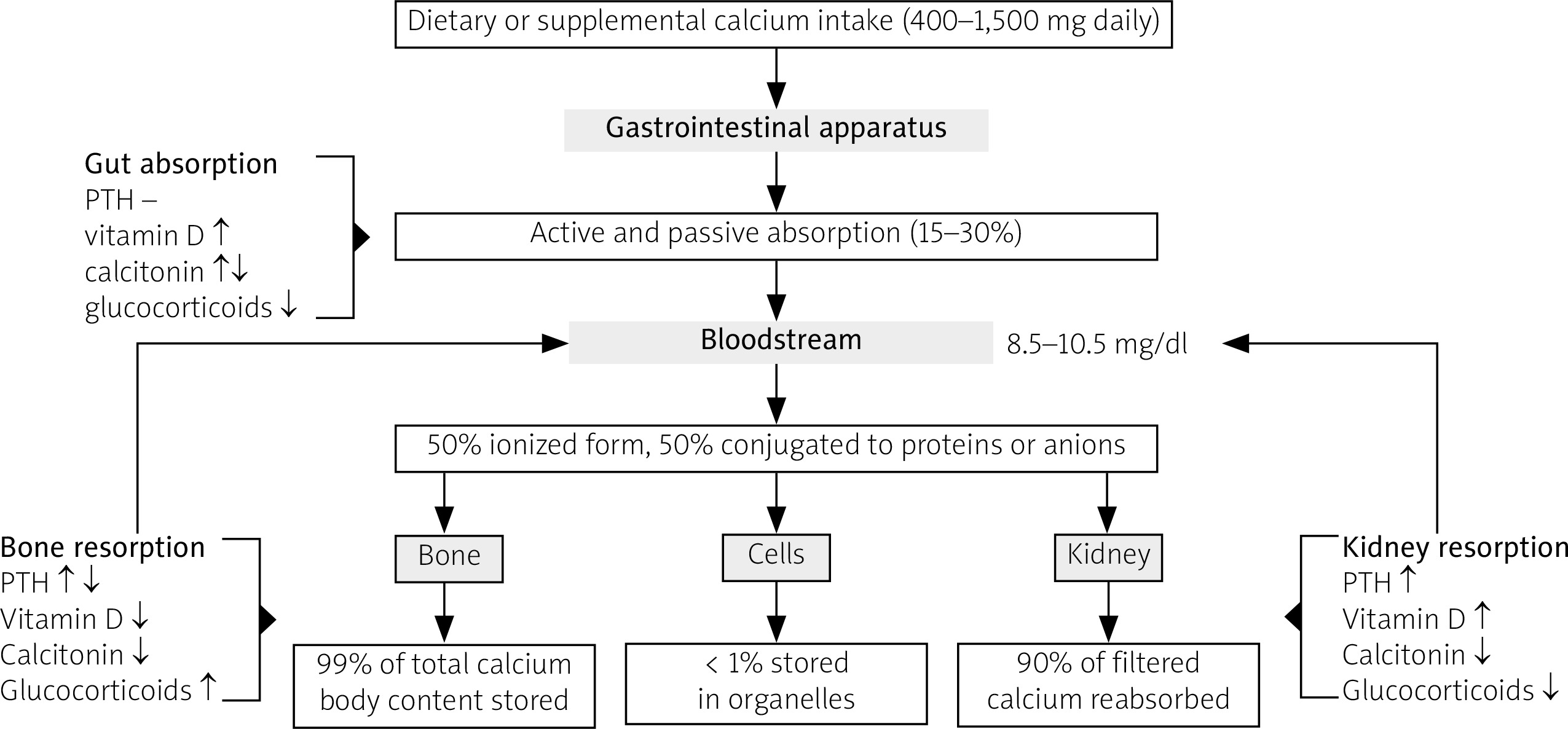
An adult human organism contains approximately 1,200 grams of calcium, of which 99% is stored in the bone, whereas the remaining percentage consists of circulating or intracellular calcium (Fig. 1). Bone exchanges with plasma about 250/300 mg of calcium in a day and contributes to maintain the normal range of calcium serum concentration.
Fig. 1
Schematic representation of calcium metabolism in physiological conditions. Blood calcium concentration derives from the fraction of calcium absorbed by the gastrointestinal tract, the fraction of calcium mobilized from skeletal deposits and the fraction of calcium reabsorbed in kidney. Each of these processes is under the control of several hormones, including PTH, vitamin D, calcitonin and glucocorticoids.
PTH – parathyroid hormone.
Within the cells, calcium acts as a second messenger and its cytosolic concentration is kept stable under a threshold of 100 nM. Slight calcium concentration variations induce the development of an electrolytic gradient that allows intracellular signaling. Sensor proteins, named stromal interaction molecule proteins (STIMs) 1, calcium channels, calcium-sequestering proteins and calcium active transporters cooperate in controlling calcium fluxes and indirectly preside over the downstream calcium-mediated cellular processes.
Extracellular calcium concentration is, instead, maintained constant thanks to the interplay of several hormones, including parathyroid hormone (PTH), 1,25(OH)2 vitamin D, calcitonin, PTH-related peptide (PTHrP), fibroblast growth factor 23 (FGF23) and klotho [2].
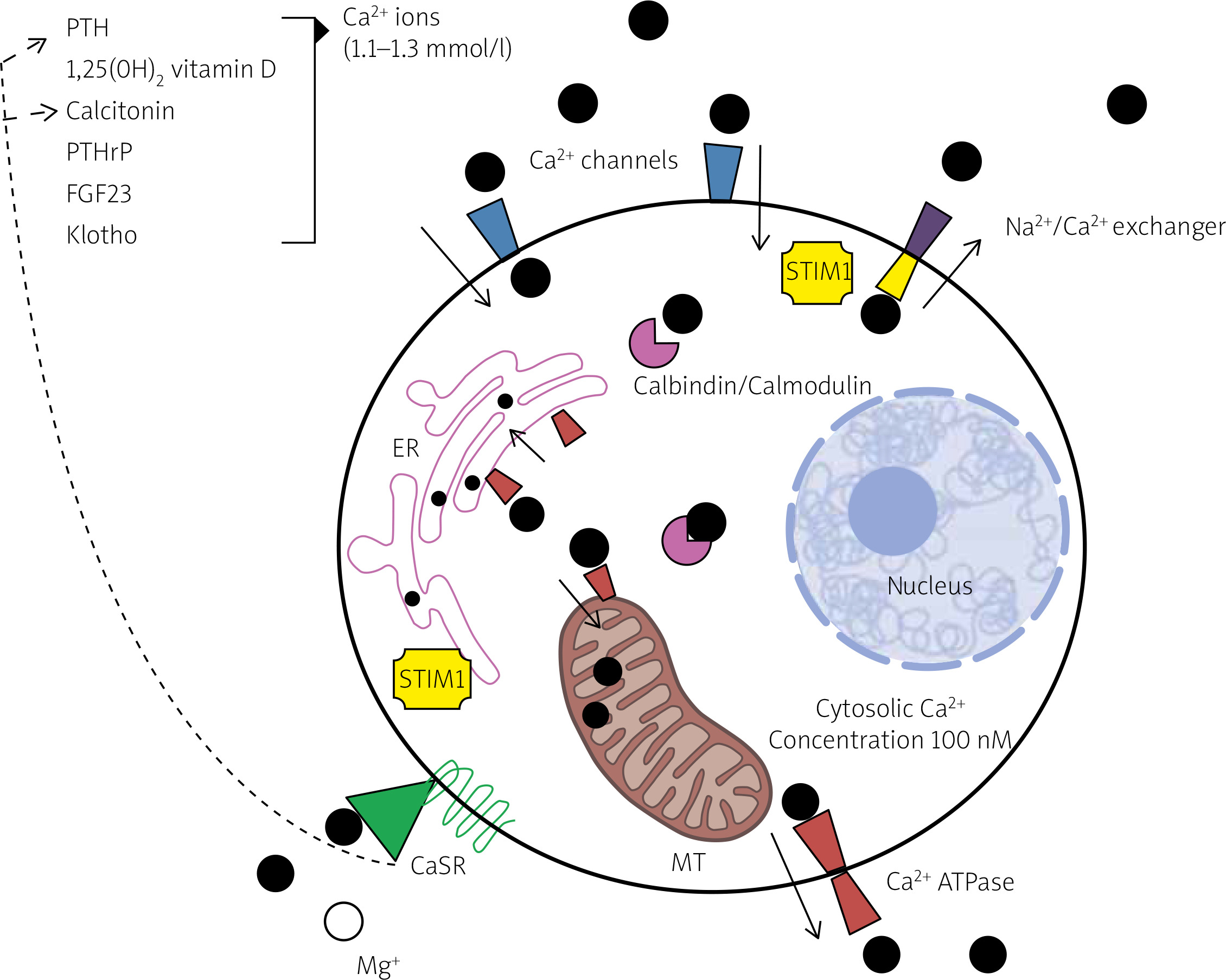
Calcium interacts as an agonist with a class of G protein-coupled receptors, known as calcium-sensing receptors (CaSRs) (Fig. 2) [11]. These receptors are present in parathyroid glands, gut, bone, kidney, calcitonin-secreting cells and other tissues. Calcium-sensing receptor activation encompasses a wide range of effects, including the induction or inhibition of hormone release (PTH, calcitonin), cellular growth or apoptosis.
Fig. 2
Regulation of intracellular and extracellular calcium homeostasis. Both the extracellular and the intracellular concentration of calcium ions are maintained in a narrow range thanks to the interplay of several hormones and an intracellular system of calcium sensing, binding and sequestering molecules. Calcium sensing receptors, expressed on plasma membrane, are sensitive to the extracellular concentration of calcium and other cations (such as magnesium) and, in case of unbalance, initiate the appropriate hormonal response by inducing or inhibiting the secretion of parathyroid hormone or calcitonin. Stromal interaction molecule protein 1 is, instead, a protein associated with endoplasmic reticulum and plasma membrane that is sensitive to intracellular calcium deposits and controls the opening of calcium channels. A stable intracellular concentration of calcium is also guaranteed by the rapid sequestration of calcium ions by the cytosolic proteins calbindin and calmodulin and the extrusion of the ion in organelles or in the extracytosolic environment through active transporters (calcium ATPases and sodium/calcium exchanger proteins).
ATPase – adenosine triphosphatase, Ca2+ – calcium, CaSR – calcium sensing receptor, ER – endoplasmic reticulum, FGF23 – fibroblast growth factor 23, Mg+ – magnesium, MT – mitochondrion, Na2+ – sodium, PTH – parathyroid hormone, PTHrP – PTH-related peptide, STIM1 – stromal interaction molecule protein 1.
Parathyroid hormone, calcitonin and vitamin D are the main factors responsible for calcium homeostasis and respond to variations in extracellular calcium concentration by modulating calcium resorption in gut, kidney and bone. Other hormones influence calcium balance and bone mineralization, including sex, thyroid and growth hormones, insulin and glucocorticoids [12].
Dietary calcium is absorbed throughout all the tracts of the intestine, by means of active (mainly in the duodenum) and passive (in ileum and colon) transportation. In the kidney, 99% of filtered calcium is reabsorbed in the proximal and distal tubule and Henle’s loop in a passive and active way.
Hormones can control calcium transportation and intracellular flux by influencing the expression of several proteins, including transient receptors potential vanilloid (TRPV)5 and TRPV6, voltage-gated L-type calcium channels, calbindins, calcium/sodium exchangers, adenosine triphosphate ATPases and claudins [2].
In physiological conditions, advisable daily calcium intake is about 400–1,500 milligrams. However, the real bioavailability of calcium is only 15–30% of the intake, although a great variability exists during growth, pregnancy and lactation [13]. Beside hormonal control, the absorption of calcium depends on age, gender, body mass, gastrointestinal pH, mucosal inflammation, dietary nutrients, gut microbiota and drugs [14].
Oxidative stress and gut inflammation may interfere with the expression of genes coding for molecules involved in both the active and passive transportation. The use of anti-oxidant drugs, as well as prebiotics and a fat-poor diet, may restore the efficiency of calcium uptake [15]. Medications, like glucocorticoids, can also influence the plasma concentration of calcium by interfering with its absorption or excretion and bone mineralization.
Calcium absorption and homeostasis in ankylosing spondylitis
Patients affected by AS often suffer from malabsorption and have macroscopic or microscopic signs of intestinal inflammation [16]. In addition, about 7% of AS patients are estimated to develop a concomitant IBD, such as Crohn’s disease or ulcerative colitis [17]. The intestinal microbiota is often altered in AS patients, with Lactobacilli less represented [4]. Gut dysbiosis can drive a local immune response, reverberating in distant sites such as joints.
In this regard, it has been shown that HLA-B27 transgenic rats reared under germ-free conditions do not develop spondyloarthritis [18] and, in humans, reactive arthritis, which belongs to the SpA group, is often the consequence of an enteric infection sustained by Salmonella, Shigella and Campylobacter spp.
The haplotype HLA-B27 may globally affect the intestinal metabolome with a loss of short-chain fatty acid (SCFA)-producing Clostridiales spp., considered at the basis of a dysregulated innate and adaptive immune response. Concomitantly, the hyper-activation of the IL-17/IL-22/IL-23 axis may further generate chronic inflammation involving distant sites, e.g. entheses and joints [4].
Both intestinal subclinical inflammation and dysbiosis may affect the absorption of calcium and other nutrients. Experiments on human cells and experimental models of IBD showed that interferon-gamma (IFN-γ) can down-regulate the expression in colon of L-type calcium channels [19], and that inflammation may overall repress the expression of TRPV6, calbindin D9K and other calcium transporters in the duodenal epithelium [20], thus reducing the amount of absorbable calcium.
Furthermore, preclinical studies in animals revealed that calcium supplementation may shift the bacterial composition of the gut microbiota towards fermenting phyla able to synthesize SCFAs [21], ultimately responsible for luminal pH acidification and increased calcium absorption.
In AS patients suffering from a concomitant IBD, calcium malabsorption might also depend on diarrhea-related hypomagnesemia, lactose intolerance and the quite common use of glucocorticoids that reduce calcium absorption in the intestine [22].
Additionally, it has been estimated that one-third of IBD subjects have an inadequate calcium intake due to dietary habits [23]. Notably, supplementation with calcium and vitamin D may be insufficient to prevent osteoporosis in IBD patients, and this may be due to the down-regulation of calcium transporters in inflamed gut mucosa as reported in preclinical studies [19, 20].
Conversely, mineral absorption in non-IBD AS patients is less clear. A case-control study showed that AS patients have a higher calcium intake compared to healthy controls, measured through the semi-quantitative food frequency questionnaire [24]. However, subclinical gut inflammation, affecting half of AS patients [16], may lead to micronutrient deficiency.
Extracellular calcium concentration has been reported within the normal range in AS subjects [25], suggesting that calcium malabsorption may be compensated by increased calcium uptake from the skeleton, partly explaining the occurrence of osteoporosis in AS subjects [20].
Additionally, a recent meta-analysis demonstrated that AS patients often have a reduced serum level of 25(OH) vitamin D, while the serum concentrations of calcium, PTH and 1,25(OH)2 vitamin D are normal [26].
Vitamin D induces gastrointestinal and renal calcium and phosphate absorption, whilst in bone it has both a catabolic and an anabolic role [12]. It is fairly well known that vitamin D has anti-inflammatory properties, and an inverse relationship between its serum level and disease activity in AS has emerged in some studies [27].
Calcium in the pathogenesis of ankylosing spondilitis
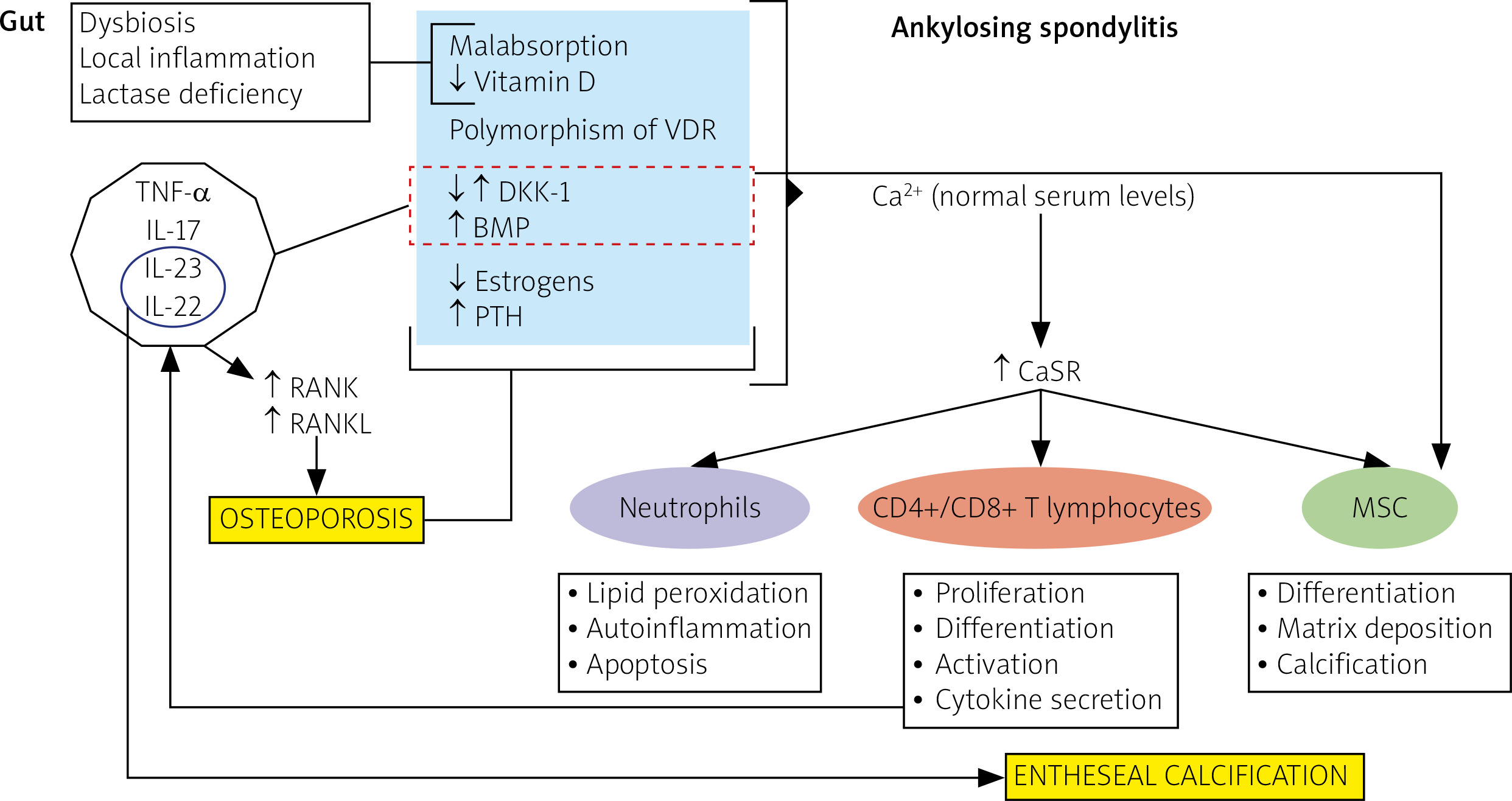
It’s presumable that an unbalanced calcium homeostasis may contribute to inflammation in the AS course, new bone tissue formation as well as bone loss. These three pathogenetic moments are separately discussed in the following subparagraphs and the hypothetical role played by calcium in the pathogenesis of AS is depicted in Figure 3.
Fig. 3
Unbalanced calcium pathways and related pathogenic mechanisms in ankylosing spondylitis. Patients affected by ankylosing spondylitis often have gut dysbiosis and inflammation, and sometimes lactase deficiency and vitamin D resistance. These conditions may induce malabsorption of calcium and vitamin D. On the other hand, the hyper-expression of pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-17, IL-23 and IL-22, may alter the balance of mediators involved in bone morphogenesis (DKK-1, BMP, RANK, RANKL). Finally, hormonal changes may further contribute to bone remodeling, leading to osteoporosis. However, in these patients, calcium, whose serum concentration is usually within the normal range, could promote the activation of immune and mesenchymal cells through increased expression of calcium sensor receptor, favoring inflammation and matrix deposition.
BMP – bone morphogenic protein, Ca2+ – calcium, CaSR – calcium sensor receptor, CD – cluster of differentiation, CGR – calcitonin gene-related peptide, DKK-1 – Dickkopf-related protein-1, IL – interleukin, MSC – mesenchymal cells, RANK – receptor activator of nuclear factor κ-B, RANKL – receptor activator of nuclear factor κ-B ligand, PTH – parathyroid hormone, VDR – vitamin D receptor gene.
Inflammation in the course of ankylosing spondylitis
Calcium may influence the immune response and repair mechanism in several ways that are summarized in Figures 4 and 5. Intracellular calcium can complex with calmodulin and activate calcium/calmodulin dependent kinases or calcineurin, which phosphorylate the inhibitor of κB kinase (IKK2) and dephosphorylate nuclear factor of activated T-cells (NFAT), respectively [1].
Fig. 4
Intracellular calcium cascade tuning the immune response. In peripheral blood mononuclear cells, intracellular calcium takes part in a phosphorylation cascade culminating in activation of the transcriptional factors nuclear factor kappa-light-chain-enhancer of activated B-cells and nuclear factor of activated T cell. The final result is transcription of the genes involved in the immune response, such as those coding for pro-inflammatory cytokines or favoring B-cell maturation. Pro-inflammatory cytokines increase, in turn, the expression of calcium sensing receptor with the subsequent repression of parathyroid hormone release and activation of 1,25(OH)2 vitamin D, which exhibits anti-inflammatory properties.
Ca2+ – calcium, CaSR – calcium sensing receptor, IL – interleukin, NFAT – nuclear factor of activated T cell, NF-κB – nuclear factor kappa-light-chain-enhancer of activated B-cells, PTH – parathyroid hormone, TNF-α – tumor necrosis factor-alpha.
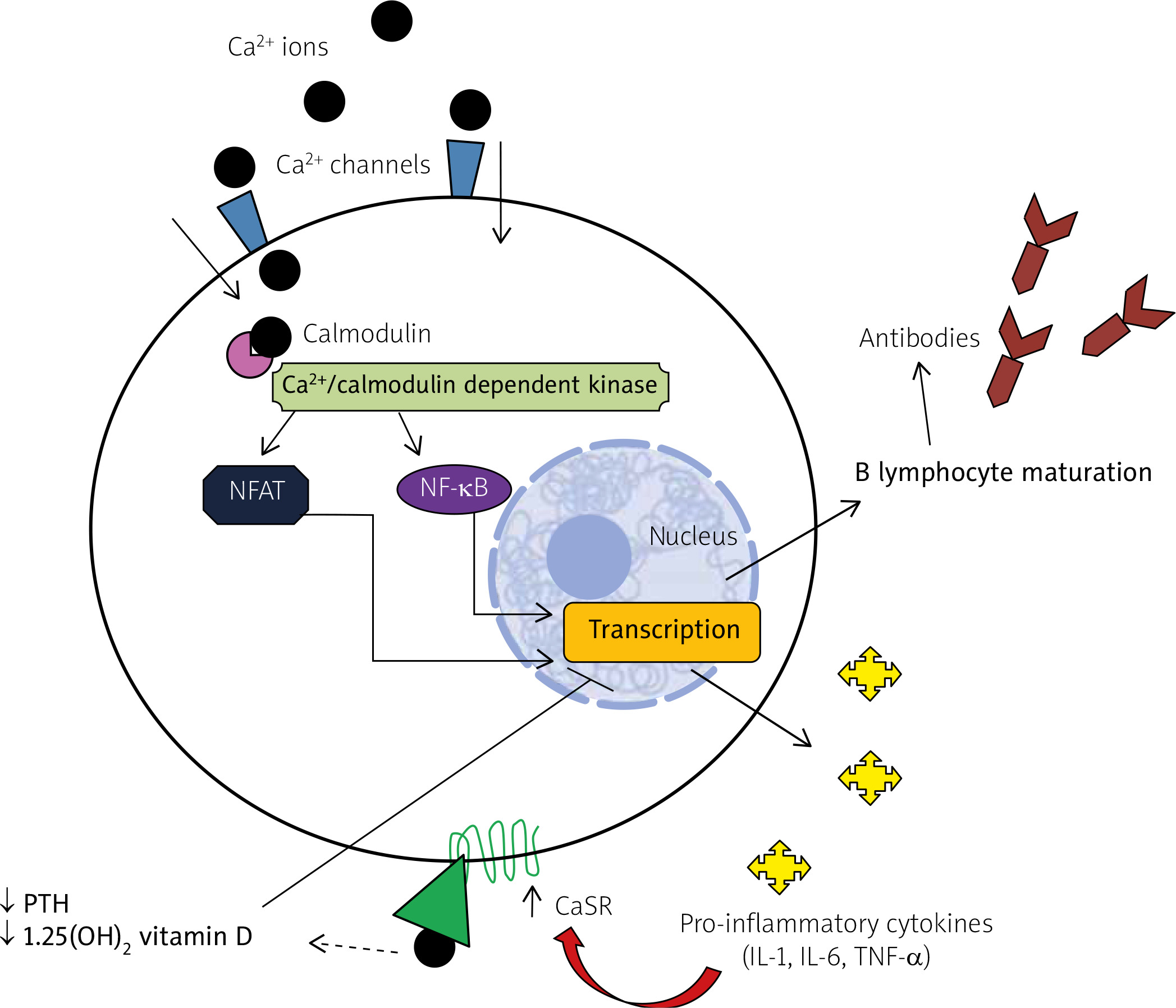
Fig. 5
The role of calcium in activation of the immune system. Calcium mediates the activation of both innate and adaptive immune cells and participates in the process of osteogenesis.
CK – cytokine.
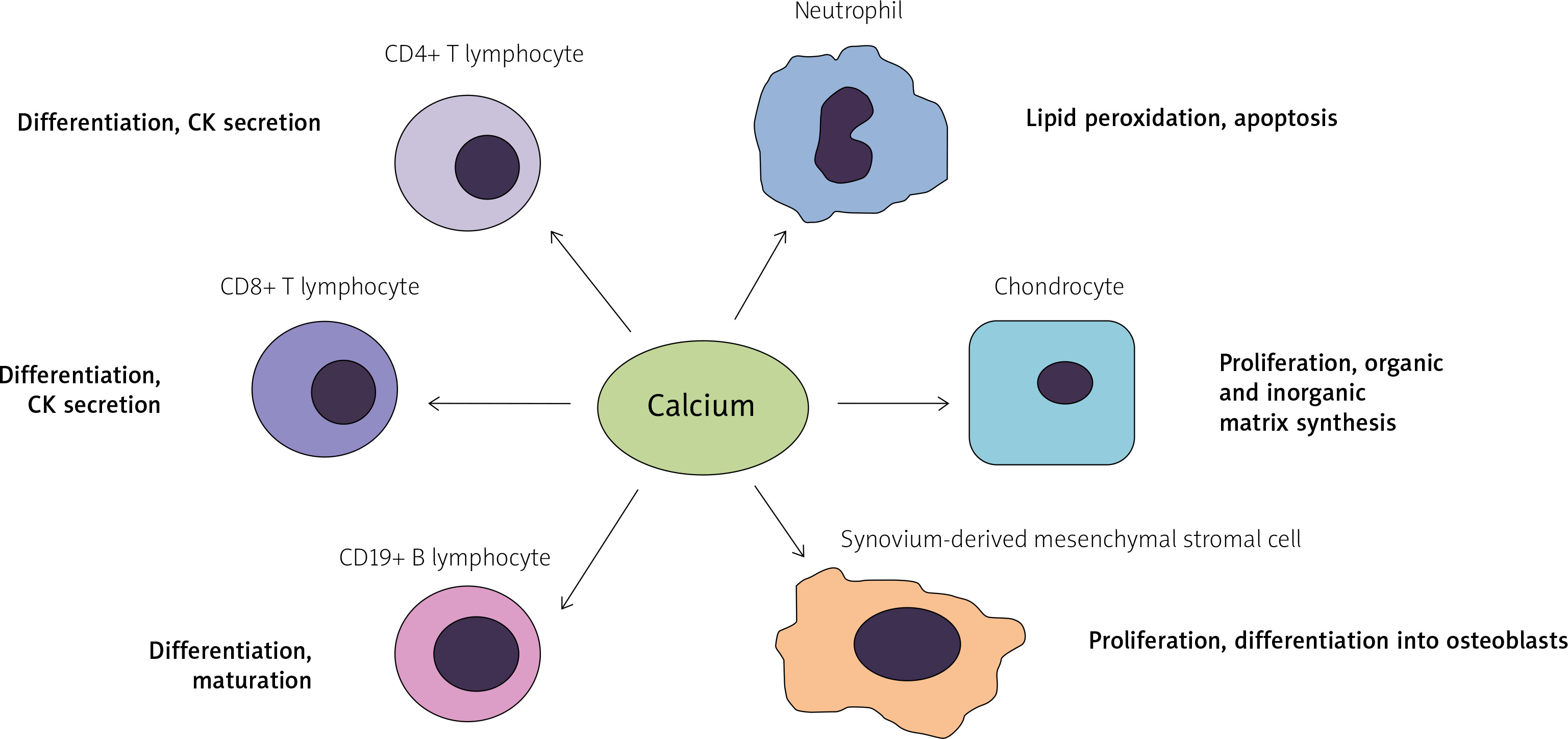
This leads to the activation and translocation of either nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) or NFAT into the nucleus, where they can control the transcription of various genes associated with proliferation and inflammation.
Additionally, the importance of calcium in inflammation has been revealed by the role that CaSRs exert in immune cells. Hypocalcemia, in fact, is often present in sepsis or other systemic inflammatory conditions, as the result of the suppression of PTH secretion.
This may depend on the aberrant expression of CaSRs on peripheral mononuclear cells (PBMCs), which use calcium for cytokine secretion and inflammasome activation [28]. Therefore, cytokines such as IL-6, TNF-α and IL-1 may indirectly influence the secretion of PTH by increasing the expression and activation of CaSRs on immune cells.
Interestingly, physiologic intracellular calcium oscillations appear typically impaired in peripheral monocytes of AS patients, and are likely associated with increased differentiation of these cells towards a dendritic or a monocytic myeloid derived suppressor cell (mMDSC) phenotype [29].
Furthermore, it has been demonstrated that intracellular calcium entrance is enhanced in neutrophils of AS patients, where it may contribute to lipid peroxidation, apoptosis, and activation of caspases 3 and 9.
Of note, the anti-TNF monoclonal antibody infliximab, used in the treatment of many rheumatic diseases including AS, may prevent calcium influx into neutrophils, thus suggesting that the opening of calcium channels may be the result of a response to cytokines [30]. Similarly, the blockade of IKCa1 lymphocyte potassium channels may display anti-inflammatory properties in AS and RA patients, preventing the differentiation of CD4+ and CD8+ T cells [31].
B lymphocytes participate to several steps of the immune response, ranging from antigen presentation to antibody and cytokine production. Nevertheless, limited data exist on the role of these cells in SpA pathogenesis.
B lymphocytes can be activated in response to intracellular calcium fluxes. After binding an antigen, the B-cell receptor (BCR) triggers a cascade of phosphorylation events; this causes the entrance of calcium ions into the cell, promoting the early differentiation and maturation of B lymphocytes [32].
Interestingly, the molecules CD19 and CD22, expressed on B cells and both targets of immunotherapy, favor and impede the calcium intracellular pathway respectively, conditioning the B lymphocyte immune response.
If intracellular calcium is of crucial importance in the activation of the immune system, the role of extracellular calcium in inflammation is less clear.
Although preclinical studies showed that calcium supplementation may worsen inflammation during sepsis [33] and that calcium-phosphate nanoparticles may activate in vitro the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome in peripheral blood monocytes [34], these data are not in line with clinical evidence [35].
Bone formation in ankylosing spondylitis
Aside from being the major component of mineral matrix in bone, calcium may also activate anabolic pathways in connective tissue cells. An in vitro model showed that the exposure of synovium-derived mesenchymal stromal cells to high calcium concentrations may cause their proliferation and differentiation into osteoblasts [36].
Likewise, another study on animal models showed that calcium levels positively affect the chondrocyte proliferation and the apposition of both organic (type X collagen) and inorganic matrix in bone during embryogenesis [37]. However, according to a recent review, calcium supplementation seems not to be associated with augmented bone mineralization in the long term [38].
In SpA, it is still unclear whether bony overgrowth at the entheseal sites does follow inflammation or rather represents a distinct and independent pathogenic event [8].
Studies in SpA animal models and human histological samples showed that entheseal calcification may be the result of endochondral and membranous osteogenesis relying on the hyper-activation of the Wingless-type MMTV integration site family (Wnt), the bone morphogenic protein (BMP) and the Hedgehog (Hh) protein anabolic pathways [8].
It has been reported that some pro-inflammatory cytokines, such as IL-1β and TNF-α, may chronically foment the Wnt pathway in chondrocytes and other cells [39]. CD4+ Th22 and Th17 cells, γδ T lymphocytes and innate lymphoid cells type III, migrating from adjacent inflamed synovial tissues to entheses [3], may induce local inflammation and subsequent calcification.
The local production of IL-17 and IL-22, together with prostaglandin E2 (PGE2), may stimulate the proliferation and differentiation of mesenchymal cells and osteoblasts [40]; conversely, TNF-α may qualitatively and quantitatively influence the local production of sclerostin and Dickkopf-related protein (DKK) 1 and perhaps prevent bone formation [41].
Dickkopf-related protein 1 is a negative regulator of the canonical Wnt signaling pathway. In AS patients, serum DKK1 levels appeared negatively associated with inflammation and radiographic progression [41, 42], and positively correlated with serum PTH levels [43].
While no evidence exists concerning the pharmacologic effect of calcium supplementation in AS bone formation, the treatment with anti-TNF drugs, counteracting local and systemic inflammation, was shown to increase the serum levels of DKK1 in AS patients, in whom it however displayed less efficient control of the Wnt signaling pathway [41].
Effect of ankylosing spondylitis on bone loss
Although ankylosing spondylitis patients are characterized by aberrant calcification of entheses, they are also affected by global osteopenia and osteoporosis, with an increased risk of fracture. Osteoporosis, due to dietary deficit, malabsorption, chronic inflammation and hypo-mobility, has been reported in more than half of AS patients [44].
Osteoporosis in AS patients is characterized by a prevalent loss of trabecular bone and is less influenced by aging, being instead associated with disease duration. Accordingly, young patients may have undetected reduced bone mineral density (BMD) with a considerable risk of fractures already evidenced at 2.5 years after AS diagnosis [44]. Moreover, these patients can also develop bone erosions in the sacroiliac joints and vertebrae [45].
Patients with nr-axSpA or early SpA also suffer from an increased risk of spinal osteoporosis, in turn correlated with inflammatory findings in lumbar MRI and serum inflammatory markers, despite normal values of serum vitamin D and PTH [46, 47].
Osteoporosis occurring in SpA is characterized by a profound alteration in bone microarchitecture and affects cortical and trabecular compartments to a different extent. These differences are even more evident considering AS and nr-ax-SpA subjects. By using high-resolution peripheral quantitative computed tomography (HR-pQCT), it has been shown that AS patients have lower cortical and trabecular volumetric (v)BMD compared to controls, and that syndesmophytes are negatively associated with trabecular vBMD and positively associated with cortical vBMD in the lumbar spine [48].
Conversely, a reduced vBMD in cortical but not in trabecular bone represents a typical early finding of nr-axSpA, although women and subjects receiving prior treatment with glucocorticoids may have a reduction in trabecular vBMD as well [49].
Therefore, it may be hypothesized that mechanical stress occurring at cortical bone may initiate the pathogenic process of local and systemic inflammation, finally ending with the resorption of trabecular bone.
In the early phase of the disease, the cytokines TNF-α, IL-1 and IL-6, released in entheses and cortical bone by monocyte-macrophage cells, may stimulate osteoblasts to express receptor activator of nuclear factor kappa-B ligand (RANKL) which, in turn, promotes the differentiation of osteoclasts from their progenitors, thus inducing bone loss.
The subsequent activation of the acquired immune system and the synthesis of other cytokines, such as IL-17, may play a double role, being associated with both bone catabolic and anabolic pathways. IL-17 may simultaneously favor the expression of RANKL, RANK and other local cytokines involved in bone resorption and promote osteoblast differentiation from mesenchymal cells [4].
The dual behavior of IL-17 may depend on the concomitant cytokine milieu (e.g. prevalence of IL-22 and IL-23 or TNF-α) and the phase of disease. Even IL-23 may favor either the production of granulocyte-macrophage colony-stimulating factor (GM-CSF) or RANKL and IL-17, which have opposite effects on bone morphogenesis.
Calcium supplementation in ankylosing spondylitis patients – the current evidence
Calcium supplementation, guaranteeing a daily dose intake within a range of 700–1,500 mg, is strongly recommended by American and European taskforces in subjects affected by osteopenia, osteoporosis or at risk of calcium deficiency [13, 50].
Although osteoporosis is a common comorbidity, its management is almost neglected in current international therapeutic guidelines for AS or SpA [5, 6]. Ankylosing spondylitis patients should be evaluated for bone loss within 1 year after AS diagnosis and those with full-blown osteoporosis should be treated with antiresorptive therapies, such as bisphosphonates or denosumab [44] plus calcium and vitamin D supplements.
Given the contribution of inflammation to AS osteoporosis, it is presumable that the use of biologic agents or even tyrosine kinase inhibitors may indirectly prevent bone resorption, whilst their role in hampering bone formation is less clear [51]. Patients with AS who were concomitantly diagnosed with IBD should be managed according to IBD therapeutic guidelines in order to prevent malnutrition and secondary osteoporosis.
Nutritional supplementation of AS patients with microscopic or subclinical colitis is unclear, due to a lack of specific studies in humans. Preclinical studies in HLA-B27 transgenic rats showed a beneficial effect of calcium supplements in reducing colic inflammation, thanks to the reinforcement of the mucosal barrier [52] that may perhaps be attributed to a rebalance in the intestinal microbiota [21].
There are several formulations of calcium: specifically, calcium carbonate, citrate, lactate and gluconate represent the supplements most commonly used in clinical practice, besides fortified food and beverages. Calcium carbonate contains 40% elemental calcium, must be administered with meals and is probably the most cost-effective formulation. Calcium citrate, containing 21% elemental calcium, is characterized by a higher solubility that renders it the calcium supplement of choice in achlorhydric, hypo-estrogenic and sideropenic patients, since its absorption is less influenced by active transport and pH [13].
Moreover, citrate contributes to bone remodeling and health [53]. Indeed, calcium citrate has more impactful effects in preventing osteopenia in both trabecular and medullar bone compared to calcium carbonate, with numerous studies confirming its better absorption, bioavailability and PTH suppression capability compared to other calcium salts [54].
Also, the formation of calcium oxalate stones seems more unlikely with the use of calcium citrate. This formulation, in fact, favors the sequestration of calcium ions and the inhibition of crystal accretion; at the same time, it augments the urinary excretion of macromolecules, such as Tamm-Horsfall protein, finally preventing the development of calcium-oxalate stones [55].
Supplementation with calcium carbonate may be associated with the risk of kidney stones in AS patients [56, 57]; in fact, those AS patients with concomitant IgA nephropathy (often due to mucosal immune system activation and gut dysbiosis) and hypercalcemia have a higher risk of hypercalciuria and kidney stones [58]. Thanks to the anti-lithogenic properties of citrate, supplementation with calcium citrate may be preferred in this subset of AS patients.
Conflicting data exist on the risk of cardiovascular events in calcium-supplemented patients [59].
Recently, the National Osteoporosis Foundation and the American Society for Preventive Cardiology stated that dietary and supplemental calcium not exceeding the threshold of 2,000 to 2,500 mg/day should be considered safe from a cardiovascular point of view [60].
Given the increased risk of cardiovascular disease in AS patients [61], the long-term cardiovascular effect of calcium salts should be addressed in further follow-up studies. Patients diagnosed with cardiovascular diseases should be carefully monitored during the treatment with calcium derivates and priority should be given to vitamin D supplementation, which showed anti-atherosclerotic and cardio-protective effects in experimental studies [62].
Based on these data, calcium supplementation should be started early in osteoporotic or osteopenic AS patients in order to prevent cortical and trabecular bone loss. In consideration of its higher efficacy than other formulations in counteracting trabecular osteoporosis and its better oral bioavailability, calcium citrate should be preferred in longstanding disease and in the case of subclinical or clinical gut involvement.
In non-osteoporotic or non-osteopenic normocalcemic AS individuals, disturbed intracellular calcium pathways may be corrected with the use of biologic agents targeting cytokines or cytokine-producer cells. By interfering with the intracellular calcium signaling, these drugs may indirectly contribute to the normalization of calcium metabolism in AS patients, preventing cytokine-driven osteopenia and calcification [51].
In fact, the use of infliximab and azathioprine and, paradoxically, of glucocorticoids showed beneficial effects on BMD in an observational cohort study on IBD patients, whereas calcium and vitamin D supplements had no significant effect [63].
Due to its anti-inflammatory role, the use of vitamin D in deficient AS subjects should be advisable along with calcium supplementation [64]. The use of vitamin D derivatives in AS patients may have several immunomodulatory effects, including the inhibition of IL-17 producing cells and the activation of IL-22 secreting cells [65]. Given the role of IL-22 in bone formation, the treatment of AS patients with supplements of vitamin D may ameliorate inflammation but possibly worsen calcification.
This effect may be further accentuated by the intestinal and renal resorption of phosphate, which may indirectly contribute to tissue mineralization through the phosphorylation of several extracellular proteins and enzymes involved in matrix remodeling [2] or directly precipitate with calcium to form hydroxyapatite crystals.
Conclusions
To conclude, it is likely that calcium plays a decisive role in the pathogenesis of AS, being involved in inflammation, bone loss and bone formation. However, very few data exist concerning the advantages and disadvantages of calcium salts in this disease.
Calcium supplementation should be mandatory in AS patients suffering from osteoporosis or osteopenia or with known risk factors of bone demineralization, such as hypo-mobility, intestinal malabsorption, comorbidities requiring prolonged use of corticosteroids and cigarette smoke.
Calcium citrate should be preferred over other formulations in the case of gut inflammation, renal calculi, and longstanding forms of disease. Calcium salts may safely be prescribed to patients diagnosed with radiographic AS and overlapping osteopenia or osteoporosis, since current scientific evidence does not support a causative role of calcium supplements in entheseal calcification.
Conversely, calcium supplements appear unnecessary in non-osteoporotic or non-osteopenic normocalcemic AS patients, who could instead benefit from the use of biologic agents. These drugs, in fact, may indirectly normalize the calcium aberrant metabolism and signaling pathways reported in AS by rebalancing the cytokine milieu, in turn associated with bone remodeling.