
Introduction
Juvenile dermatomyositis (JDM) is a heterogeneous autoimmune inflammatory myositis with a symmetrical proximal muscle weakness and a characteristic rash [1–3]. Juvenile dermatomyositis is classified as a systemic immune mediated vasculopathy; therefore the gastrointestinal tract, the respiratory system and other organs and systems may also be involved in the pathologic process [1, 4].
Juvenile dermatomyositis is a rare disease; its annual incidence ranges from 2 to 4 cases per million and depends on the race [5]. Girls are affected more often than boys [1, 5]. The disease begins at the age of 4 to 10 years, with an earlier onset in females [2].
Genetic predisposition in combination with the influence of environmental factors, especially exposure to ultraviolet light, is important for the disease development [6]. Infections are also crucial, in particular Coxsackie virus, influenza virus, parvovirus, hepatitis B virus, group A streptococcus, toxoplasma and Borrelia [7, 8]. Studies and observations in recent years have shown the possible role of SARS-CoV-2 infection as a trigger for dermatomyositis [9, 10].
Clinical guidelines for the diagnosis of JDM are based on the criteria of Bohan and Peter, which include: typical skin signs (heliotropic rash, Gottron’s papules and sign); symmetrical progressing proximal muscle weakness; elevation in serum of skeletal muscle enzymes: creatine phosphokinase (CPK), lactate dehydrogenase (LDH), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and aldolase; myopathic abnormalities on electromyography (EMG) and biopsy data [11]. Three or four criteria (plus the rash) are needed for the definite diagnosis of dermatomyositis (DM); two criteria (plus the rash) are needed for the probable diagnosis and one criterion (plus the rash) is needed for the possible diagnosis of DM [11]. The EULAR/ACR developed the classification of “definite”, “probable”, and “possible” idiopathic inflammatory myopathies (IIM), including juvenile IIM, which allows better recognition of IIM [12].
Juvenile dermatomyositis is characterized by variable presentation and phenotypes which may make it difficult to recognize in the early stages [4]. Cutaneous manifestations may not be very specific in the early stages and appear earlier than myopathy [13]. Detecting myositis autoantibodies and using magnetic resonance imaging (MRI) may improve the possibility of JDM diagnosis [13]. However, phenotypic overlap may exist with other systemic inflammatory diseases. The average time between the appearance of the first symptoms and confirmation of the diagnosis is 6 months, and ranges from 5 weeks to 2 years [2]. Therefore, the analysis of clinical features, laboratory data and myositis autoantibodies in altered environmental conditions is of interest and helps to identify problems and plan further research, which may change the understanding of the disease and its prognosis.
Objective and methods
The objective of this article is to present and analyze the clinical and autoantibody phenotypes of JDM in patients from one center in Ukraine.
Methods: case study presentation as a basis for discussion, the search of MEDLINE (PubMed) and Scopus database in the subject of JDM, using the combination of words “juvenile dermatomyositis”, “clinical course”, “clinical phenotype”, “diagnosis”, and “myositis autoantibodies”; discussion of the problem based on the clinical cases and cited articles. We used relevant full-text articles in English published between January 2012 and April 2022. We also used some basic articles that contained classification, diagnostic and treatment approaches that were published earlier. Studies with results related to the clinical presentation, diagnosis, clinical and autoantibody phenotypes in patients with JDM were selected for the analysis.
Results
We present the cases of four patients (three girls and one boy) with JDM. We observed these patients from 9 months to more than 2.5 years. In all these cases the diagnosis was based on the criteria of Bohan and Peter [11] and also was classified with IIM according to the EULAR/ACR criteria [12]. Informed consent was obtained from all patients and/or their parents.
Case descriptions
Case 1
A 9-year-old boy was admitted to the rheumatology department of the Regional Children’s Hospital with complaints of pain in the knee joints, muscles of the proximal upper and lower extremities, muscle weakness, inability to tie shoelaces, difficulty climbing stairs, muscle tenderness, low appetite, and weight loss.
Arthralgia was observed for about 2 months; it had a progressive course. At the first visit to the doctor after 6 weeks from the onset of symptoms, arthritis of both knee joints was detected, and synovitis was observed on ultrasound. Juvenile idiopathic arthritis (JIA) was suspected. The prescribed non-steroidal anti-inflammatory drug (NSAID) had a slight positive effect, followed by pain in the muscles of the thighs and shoulders, soreness and limited movement in the hip and wrist. Myositis was suspected and the boy was referred to hospital.
Baseline characteristics are summarized in Table I. The skin was moderately pale. A slight purple discoloration of the eyelids, pale Gottron’s papules over the elbows, metacarpophalangeal, proximal interphalangeal, and knee joints, the shawl sign, mechanic’s hands, and moderate dryness of other parts of skin were seen. Cardiac and respiratory changes were not observed. The heart rate was 78 per minute. Blood pressure was 90/60 mm Hg. Symmetrical pain and restriction of movement in the knee, hip and wrist joints, and pain on palpation of the shoulder muscles and the thighs were observed.
Table I
Baseline characteristics, clinical features and laboratory indicators in presented cases
| Sign | Case 1 | Case 2 | Case 3 | Case 4 |
|---|---|---|---|---|
| Gender | Male | Female | Female | Female |
| Disease onset (time) | July 2019 | May 2020 | October 2020 | August 2021 |
| Date of diagnosis | September 2019 | February 2021 | November 2020 | September 2021 |
| Age at diagnosis, years | 9 | 7 | 10 | 9 |
| Disease onset (course)* | Subacute | Insidious | Acute | Subacute |
| Weight [kg] (z-score) | 24.5 (–1.12) | 25 (0.65) | 25 (–1.74) | 40 (1.12) |
| Height [cm] (z-score) | 128 (–1.02) | 130 (1.64) | 135 (–0.62) | 150 (2.04) |
| BMI (z-score) | 15.0 (–0.81) | 14.8 (–0.42) | 13.7 (–1.94) | 17.8 (0.46) |
| Main clinical manifestations | ||||
| Objective symmetric muscle weakness | Yes | Yes | Yes | Yes |
| Heliotrope rash | Yes | Yes | Yes | Yes |
| Gottron’s papules | No | Yes | No | Yes |
| Gottron’s sign | Yes | Yes | Yes (mild) | Yes |
| Other clinical manifestations | ||||
| Arthritis | Yes | Yes | No | No |
| Polyarthralgia | Yes | Yes | No | Yes |
| Muscle pain | Yes | Yes | Yes | Yes |
| Muscle tenderness | Yes | No | No | No |
| Raynaud’s phenomenon | No | Yes | Yes | Yes |
| Shawl sign | Yes | No | No | No |
| Periorbital edema | No | Yes | Yes | Yes |
| Mechanic’s hands | Yes | No | No | No |
| Unexplained fever | No | No | Yes | Yes (shortly) |
| Laboratory indicators (reference) | ||||
| ESR [mm/h] (0–15) | 8 | 4 | 33 | 44 |
| CRP [mg/l] (< 5 mg/l) | 0.1 | 0.79 | 92.16 | 0.95 |
| ALT [U/l] (< 37 U/l) | 10.2 | 40.3 | 14.7 | 163.3 |
| AST [U/l] (< 40 U/l) | 34.1 | 60.5 | 24.3 | 490.0 |
| CPK [U/l] (< 154 U/l) | 264.5 | 281.9 | 20.9 | 4067 |
| LDH [U/l] (120–300 U/l) | 307.3 | 433.3 | 269.9 | 1036 |
| ANA, titer (< 1:100) | < 1:100 | < 1:100 | < 1:100 | < 1:100 |
| Myositis autoantibodies (IgG) | ||||
| Anti-Mi-2 | Negative | Negative | Threshold | Positive |
| Anti-Кu | Threshold | Positive | Negative | Threshold |
| Anti-PM/Scl complex | Positive | Negative | Negative | Negative |
| Anti-histidyl-tRNA synthetase (Jo-1) | Negative | Negative | Negative | Negative |
| Anti-threonyl-tRNA synthetase (PL-7) | Positive | Positive | Positive | Positive |
| Anti-alanyl-tRNA synthetase (PL-12) | Positive | Threshold | Negative | Negative |
| Ring-dependent E3 ligase (Ro-52) | Negative | Negative | Negative | Positive |
| EULAR/ACR classification criteria [12] | ||||
| Score range | 10.1–10.1 | 12.2–14.1 | 8.1–10 | 12.2–14.1 |
| Probability | 99–99% | 100% | 94–99% | 100 % |
| Classification | Definite IIM | Definite IIM | Definite IIM | Definite IIM |
| Subgroup | JDM | JDM | JDM | JDM |
* Onset and progression of the first symptoms to the full disease presentation: acute (days to 2 weeks); subacute (> 2 weeks to ≤ 2 months); insidious (> 2 months to years) [12].
ANA – antinuclear antibodies, ALT – alanine aminotransferase, AST – aspartate aminotransferase, BMI – body mass index, CPK – creatine phosphokinase, CRP – C-reactive protein, ESR – erythrocyte sedimentation rate, IIM – idiopathic inflammatory myopathies, JDM – juvenile dermatomyositis, LDH – lactate dehydrogenase.
We detected elevation serum CPK and LDH. Anti-threonyl-tRNA synthetase (PL-7), anti-alanyl-tRNA synthetase (PL-12) and anti-PM/Scl complex autoantibodies were revealed (Table I). The EMG showed reduction of muscle function by 60–65% below normal, MRI of the soft tissues of the thigh confirmed inflammatory changes in the muscles, ultrasound of the joints revealed signs of bursitis and synovitis of both knee joints and signs of arthritis of the right hip joint.
Taking into account progressive muscle weakness, dermatologic features (purple erythema of the eyelids, Gottron’s papules, mechanic’s hands, shawl sign), elevated levels of muscle enzymes, detecting myositis autoantibodies, EMG abnormalities, and inflammatory muscle changes on MRI, JDM was diagnosed.
The patient was treated with glucocorticosteroids (GCs; oral methylprednisolone [MP] 2 mg/kg b.w./day), subcutaneous methotrexate (MTX) 15 mg/m2 weekly and adjunctive medicines (folic acid the day after MTX, ranitidine, calcium/vitamin D). He responded well to the prescribed treatment. The boy has been followed up for 2.5 years and no exacerbations were observed for this period. Currently the patient is active and continues MTX therapy.
Case 2
A 7-year-old girl was admitted to the rheumatology department of the Regional Children’s Hospital with complaints of severe weakness, difficulty walking, climbing stairs, and severe erythematous rash on the face and upper extremities. It was found out that the rash on the face had been observed for about 10 months. The girl was examined and treated by a pediatrician and an allergist, as the rash was considered allergic. Antihistamines and topical therapy did not work and after the summer the rash intensified. Severe weakness and gait disturbances began a month before admission to the hospital.
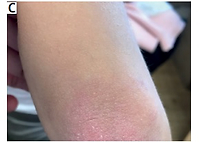
The patient’s baseline characteristics and main clinical data are shown in Table I. Periorbital edema, heliotrope rash (Fig. 1 A), Gottron’s papules and sign, Raynaud’s phenomenon (Fig. 1 B–D), and erythematous rash on the shoulder (Fig. 1 E) were seen. Examination revealed decreased muscle strength of the proximal muscles of the lower and upper extremities. The liver was +1.5 cm below the edge of the right costal arch. There was no evidence of other organ involvement.
Fig. 1
Case 2. Appearance of the patient at presentation. Images showing heliotrope rash and periorbital edema (A), Gottron’s papules and sign over the knees, elbow and hands (B–D), Raynaud’s phenomenon (D), erythematous lesion on the shoulder (E). Images are used with mother’s and child’s consent.
Laboratory examination detected elevation in muscle enzymes (AST, CPK, LDH) and the presence of myositis autoantibodies (Table I). Ultrasound of the knee joints showed synovitis, bursitis on the left, synovitis on the right. Electromyography revealed a decrease in muscle function by 60–30%. Other inflammatory systemic diseases of connective tissue, especially systemic lupus erythematosus, were ruled out.
Pulse therapy with intravenous MP (i.v. MP) followed by oral MP 1 mg/kg b.w./day and subcutaneous MTX 15 mg/m2 weekly was administered. Adjunctive therapy included folic acid, omeprazole, vitamin D, and calcium. At the time of discharge, the patient’s condition improved, the rash decreased slightly, muscle strength improved significantly, and AST, CPK, and LDH normalized. However, despite the rapid positive dynamics of the myopathic syndrome, skin rashes regressed slowly. Currently the patient continues MTX treatment.
Case 3
A 10-year-old girl was admitted to the rheumatology department of the Regional Children’s Hospital with complaints of severe weakness; fever up to 38°C; rash on the face, torso, and hands that was worsened by fever; headache and leg muscle pain. These symptoms had been observed for about 2 weeks. The NSAID treatment at the outpatient stage was without a pronounced response, so the girl was hospitalized in the infectious disease department.
Laboratory testing revealed anemia (hemoglobin 90–106 g/l), lymphopenia (1,159/µl), an erythrocyte sedimentation rate (ESR) of 28 mm/h, and C-reactive protein (CRP) of 20.1 mg/l. Polymerase chain reaction (PCR) and serology for SARS-CoV-2 infection were negative. Antibacterial therapy was without positive effects; on the contrary, the child’s condition worsened. Sternal puncture was performed to rule out oncohematological pathology. Chest computed tomography was conducted and no changes were found. The girl was transferred to the rheumatology department with suspected systemic inflammatory connective tissue disease.
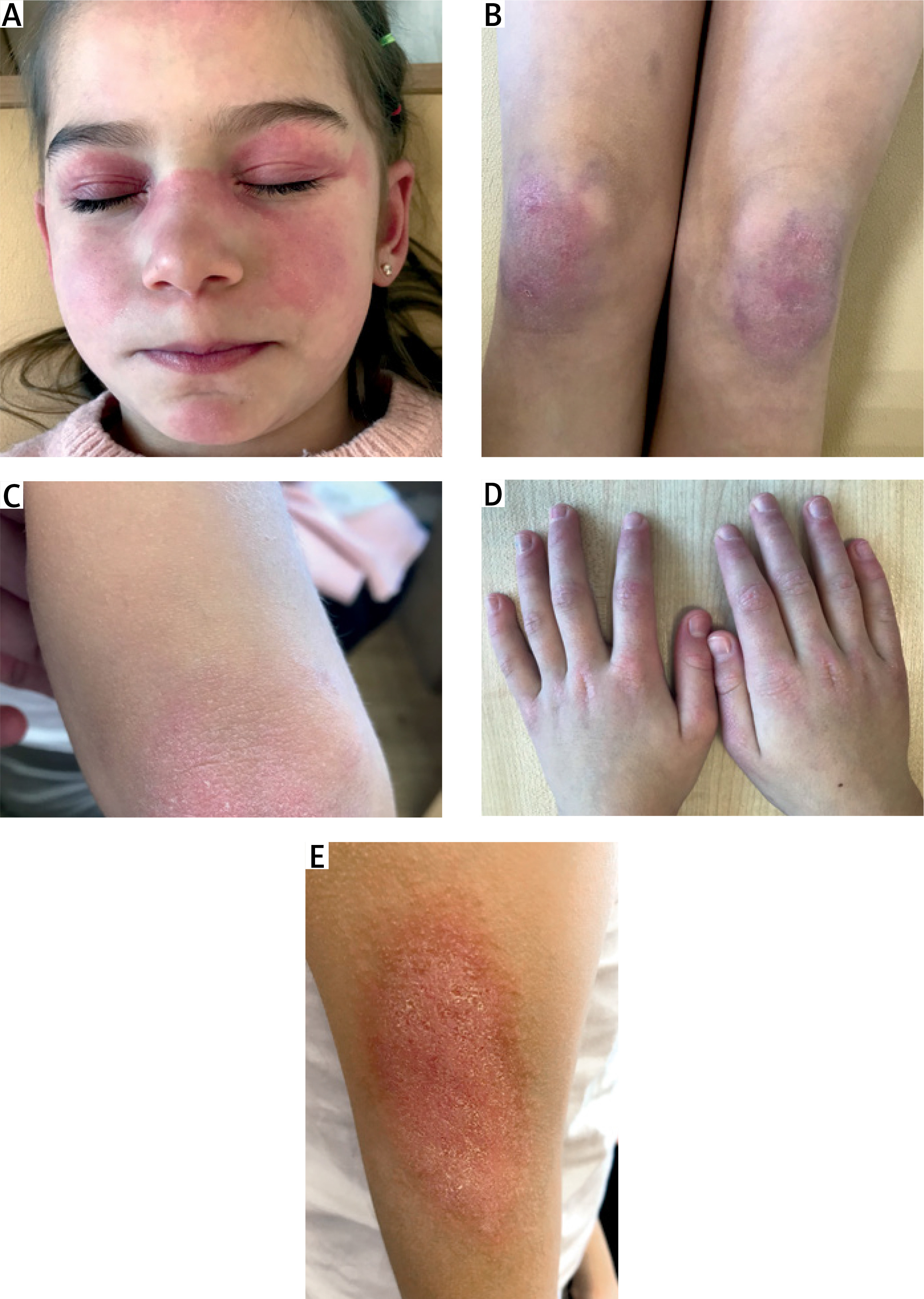
Baseline data and main clinical manifestations are summarized in Table I. Examination revealed pale skin, periorbital heliotrope rash and edema (Fig. 2), mild Gottron’s sign over the elbows, erythematous rash on the face, torso, and palms, which intensified at the height of fever; decreased muscle strength in the proximal muscles of the lower and upper extremities. The liver was +1.5 cm below the edge of the right costal arch.
Fig. 2
Case 3. Appearance of the patient at presentation. Images showing heliotrope rash and periorbital edema. Image is used with mother’s and child’s consent.
Complete blood count (CBC) revealed anemia (hemoglobin – 96 g/l), leukocytosis (22,500/µl), neutrophilia (17,600/µl) with a shift to the left, and relative lymphopenia (13%). An increase in the level of systemic inflammation data (ESR, CRP) and the presence of myositis autoantibodies was seen (Table II). However, the indicators CPK, LDH, AST were within normal limits. Electromyography showed decreased muscle function. There was no evidence of lung damage.
Table II
Association of myositis autoantibodies with frequency and clinical phenotype of IIM
| Myositis autoantibodies | Frequency | Clinical phenotype | |
|---|---|---|---|
| Adults | Children | ||
| Myositis-specific autoantibodies: | |||
| Anti-Mi-2 | 5–50% [25] | 4–10% [3, 21, 22] | Lower organ involvement, severe myopathy, good response to standard treatment [3] Protective effect: more likely drug-free remission [19] Increased risk of cancer-associated myositis and cancer [25] |
| Anti-synthetase syndrome | |||
| Anti-histidyl-tRNA synthetase (Jo-1) | 15–30% [25] | Collectively < 5% [3] | More severe muscle involvement [19] |
| Anti-threonyl-tRNA synthetase (PL-7) | Collectively 10–20% [3, 25] | More prevalent and severe lung involvement [19] Gastrointestinal complication [23] | |
| Anti-alanyl-tRNA synthetase (PL-12) | More prevalent and severe lung involvement [19] | ||
| Ring-dependent E3 ligase (Ro-52) | n.a. | 14% [21, 26] | Earlier development of mechanic’s hands, specific skin signs, arthritis [19] Higher cancer risk and more severe muscle and joint involvement [24] More likely ILD; chronic disease course, more severe disease, a poorer prognosis [26] |
| Myositis-associated autoantibodies: | |||
| Anti-Кu | 1.5% [25] | < 1% [21] | Connective tissue disease overlap; arthritis; Raynaud’s; ILD [25] |
| Anti-PM/Scl complex | 7.3% [25] | 4–5% [3, 21] | Connective tissue disease overlap; cutaneous involvement (mechanic’s hands, rash); Raynaud’s; ILD [25] |
Treatment included dexamethasone intravenously at a dose of 2 mg/kg b.w./day for prednisolone followed by oral MP at a dose of 1 mg/kg/day, MTX 15 mg/m2/week, and adjunctive therapy (folic acid, omeprazole, vitamin D, calcium).
The patient responded to the treatment, the girl’s condition improved, the temperature normalized, the rash significantly decreased, muscle strength improved, and inflammation indicators decreased. There was a complete regression of skin changes, myopathic syndrome, and normalization of laboratory parameters (ESR, CRP). However, after 6 months of therapy with MTX and maintenance doses of MP, an increase in transaminases, mainly ALT up to 400 U/l, was observed. Probable causes of elevated enzymes (viral hepatitis B and C, cytomegalovirus infection, Epstein-Barr infection, autoimmune hepatitis) were ruled out.
Case 4
A 10-year-old girl was admitted to the rheumatology department of the Regional Children’s Hospital because of the rash on her face, upper and lower extremities, severe weakness, back pain, muscle aches, and swelling around the eyes. It was found out that the symptoms had been observed for about 1 month. There was a short-term fever up to 38°C at the onset of the disease followed by rashes on the face and hands of both extremities. Weakness, and pain in the joints and skeletal muscle gradually developed.
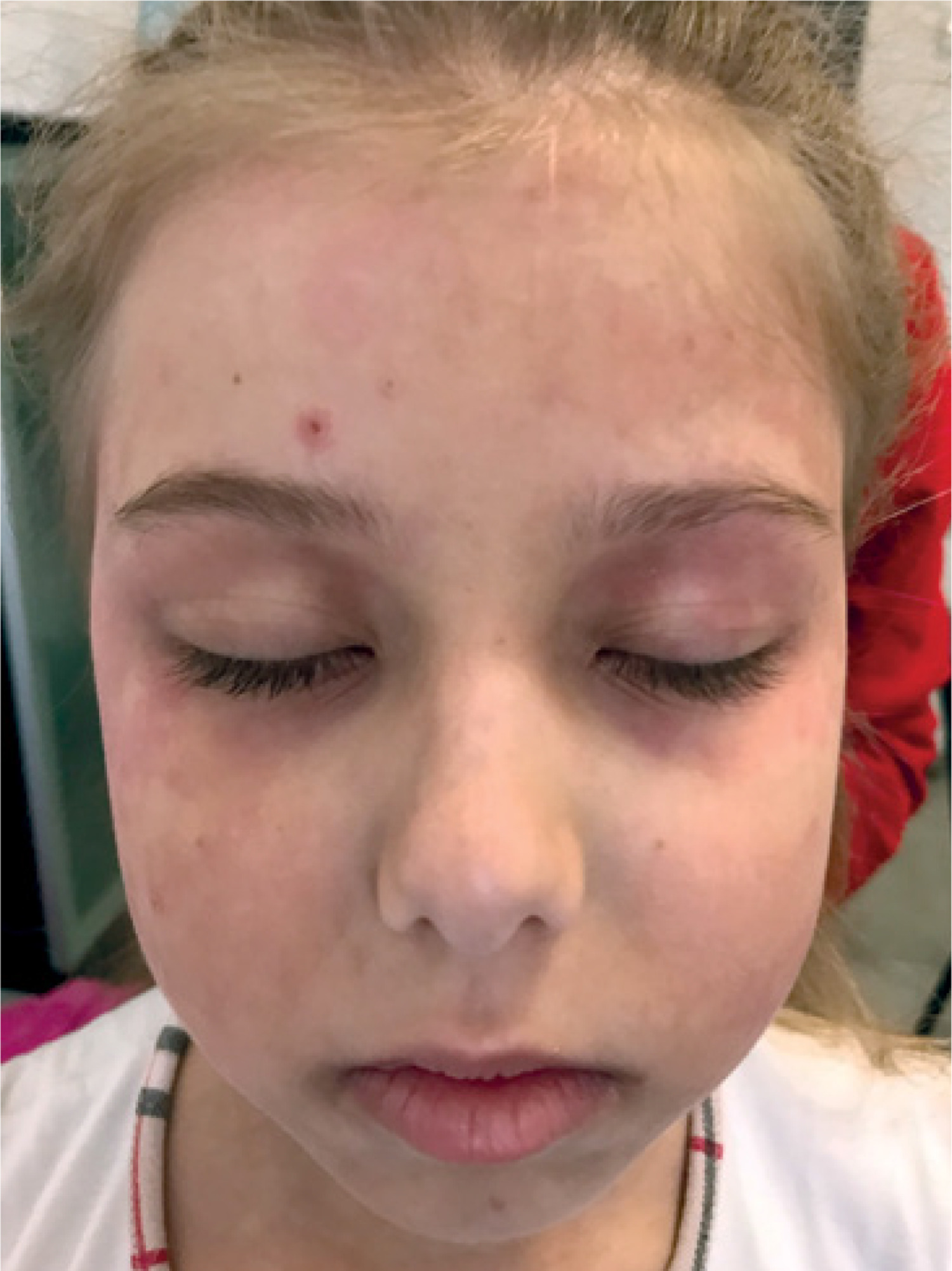
The patient’s baseline characteristics and main clinical manifestations are presented in Table I. On examination, periorbital edema, the heliotrope rash on eyelids, purple erythema on the cheeks, forehead (Fig. 3 A), Raynaud’s phenomenon, Gottron’s papule and sign over the elbows, knees, and finger joints (Fig. 3 B), and erythematous patches over the shoulder (Fig. 3 C) were observed. There was no evidence of cardiac or respiratory system involvement. The liver was +1 cm below the edge of the right costal arch.
Fig. 3
Case 4. Appearance of the patient at presentation. Images showing heliotrope rash and periorbital edema (A); Gottron’s papules and sign over the knees (B); erythematous patches over the shoulder (C). Images are used with mother’s and child’s consent.
Mild lymphopenia (1,020/µl), elevated ESR, significant changes of muscle enzymes and myositis autoantibodies (Table I) were detected.
Pulse i.v. MP 15 mg/kg/dose for 3 consecutive days followed by oral MP 1 mg/kg b.w./day and subcutaneous MTX 15 mg/m2 weekly was prescribed. Adjunctive therapy included folic acid, pump inhibitors, vitamin D and calcium.
The patient responded well to the treatment. The patient’s condition improved, the rash decreased, and muscle weakness gradually resolved. There were also positive changes in laboratory indicators, although CPK, LDH, and AST completely normalized after 2 months of treatment. During the observation, remission was achieved. The girl is currently receiving a maintenance dose of MP and MTX.
Discussion
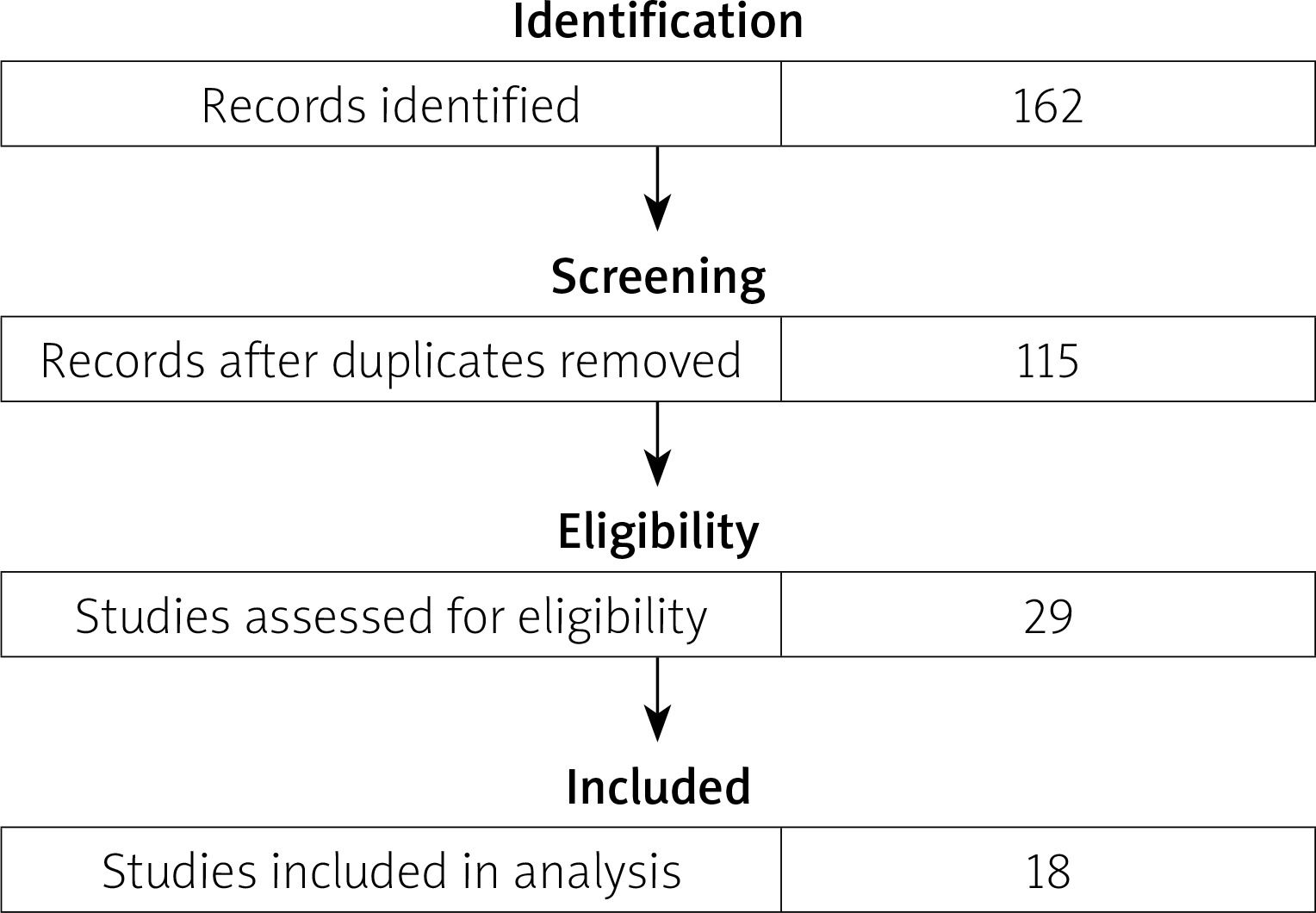
The results of the literature review are presented in a flow diagram (Fig. 4). The search resulted in 135 records in PubMed and 27 records in Scopus. Overall, 47 records were excluded because of duplicates. After screening titles and abstracts, 86 records were excluded: 12 articles not available in English, 38 review articles, 36 – irrelevant study topic. After assessing full-text articles for eligibility, 18 studies were included in the comparative analysis. Studies with duplicate data, no full text available, or an irrelevant study topic were excluded.
Herein we present and analyze four cases of JDM in children who have been hospitalized in one regional center during the last 3 years. In the first case the disease manifested before the COVID-19 pandemic period, whereas three other patients presented during the 1.5 years of the COVID-19 pandemic. It should be noted that in the pre-pandemic period we recorded an average of 1 patient with JDM for 2–3 years, which corresponded to the incidence in other populations, given the number of children in the region (194 thousand) [5]. The role of SARS-CoV-2 infection and increase in the incidence of systemic connective tissue diseases, including JDM during the COVID-19 pandemic, is also indicated in other studies [9, 10, 14].
Three out of the four presented patients were girls, which coincided with other publications indicating a predominance of females [1, 2]. The age of onset ranged from 7 to 10 years, which was similar to another study [2].
According to Bohan and Peter criteria, the diagnosis of JDM should include specific dermatological signs, symmetrical proximal muscle weakness, elevated levels of skeletal muscle enzymes (CPK, LDH), muscle biopsy and EMG abnormalities [11]. However, pediatric experts and dermatologists suggest that in patients with typical skin rashes, JDM can be diagnosed without muscle biopsy, and new classification criteria have been developed without these data [12]. However, where a pathognomonic skin rash is not observed, a muscle biopsy is required for diagnosis [12]. The presented patients, despite the different onset of the disease, had a characteristic skin rash and symmetrical muscle weakness on admission, which facilitated the diagnosis at this stage without muscle biopsy. All patients were diagnosed with IIM according to the EULAR/ACR criteria using the online web calculator (www.imm.ki.se/biostatistics/calculators/iim) [12] and the definition of IIM. The subgroup JDM was confirmed in all patients with probability of 94–100% (Table I). Disease onset was also determined according to these criteria. Acute onset was observed in one patient, subacute in two cases and insidious in one case (Table I).
Other clinical signs included muscle pain (4/4), Raynaud’s phenomenon (3/4), periorbital edema (3/4), and polyarthralgia (3/4). Arthritis and unexplained fever were presented in 2/4 cases, muscle tenderness in 1/4 case, and the shawl sign in 1/4 case (Table I). We did not find evidence of dysphagia, esophageal dysmotility, lung or cardiac involvement, or calcinosis in the presented patients, although scientific data have been reported on the possibility of such symptoms in children with JDM [2, 8, 12, 15]. Interstitial lung disease (ILD) and cardiac involvement are very rare (2–6%) in the onset of JDM [13, 16], whereas calcinosis is observed more often in children than in adult [13].
Among the laboratory indicators, increased ESR was detected in 2 cases, while a significant increase in CRP was seen in only one case. Elevated serum levels of CPK and LDH were observed in 3 out of 4 cases. The ALT and AST were increased in 2 out of 4 cases. Bohan and Peter also pointed out that “serum enzymes are not infallible guidelines” [11] and in some cases they may be normal despite active myositis [13, 17].
Despite the similarity of certain clinical and laboratory signs, each of the presented cases had its own pattern in the onset, which in some cases led to late diagnosis.
In the onset of the disease in the first case the patient presented with polyarthralgia and arthritis, without skin manifestations, so juvenile idiopathic arthritis (JIA) was initially suspected. Muscle tenderness was initially regarded as the morning stiffness that is typical for JIA. Later, when muscle weakness and muscle pain developed, JDM was suspected. Heliotrope rash in this patient was less pronounced in comparison to other patients. In the second case, the skin lesions were typical, significant, but the absence of myopathic syndrome over six months from the onset of the disease led to the late JDM diagnosis. The third case was the most controversial, as it had an acute onset with fever, increased inflammation indicators (CRP, ESR), leukocytosis, neutrophilia, and lymphopenia, which led to admission to the infectious disease department, where bacterial infections and COVID-19 were ruled out. Persistent fever for more than 2 weeks, increased rash on the background of fever, and severe inflammation required exclusion of systemic JIA [18]. The patient also presented with characteristic clinical signs of JDM (dermatologic features and muscle weakness), although skeletal muscle enzymes were not elevated. The last case had the most typical course in the onset of the disease with pronounced dermatological features and significant laboratory changes (elevated levels of muscle enzymes CPK, LDH, ALT, AST) (Table I).
Detection of myositis autoantibodies is a very useful tool for JDM diagnosis as they can predict disease course, systemic involvement, and malignancy risk [3, 8, 13, 15, 19, 20]. The frequency and association of myositis autoantibodies with clinical phenotype of IIM are presented in Table II. Myositis-specific or associated autoantibodies were identified in 60–95% of JDM patients [3, 21, 22]. They were found in all our patients. Children with classic JDM often presented with such types of myositis autoantibodies as anti-Mi-2 (4–10%), anti-NXP2 (15–23%), anti-TIFγ (18–32%) and anti-MDA5 (7–38%) [3, 20–22]. Anti-MDA5 antibody positive JDM is associated with arthritis, weight loss, adenopathy, less severe myositis, a rapid response to steroids and less frequent flares compared to anti-TIFγ antibodies [23]. We were able to detect anti-Mi-2 autoantibodies among those mentioned above. Anti-Mi-2 autoantibodies are associated with severe pronounced onset of JDM but a good response to therapy and good prognosis (Table II). Only in one out of the four presented patients were anti-Mi-2 autoantibodies detected (case 4). This girl had a highly pronounced onset of JDM.
Among myositis autoantibodies are anti-synthetase autoantibodies (anti-Jo-1, anti-Pl-7, anti-Pl-12), which are under the umbrella of anti-synthetase syndrome (ASyS) [24]. This syndrome is common in adult patients with myositis but is rare in children [3]. Less than 5% of children with JDM presented with ASyS [8, 21]. In recent years, ASyS has been important in the diagnosis of connective tissue diseases and IIM, including JDM, as well as correlated with distinct DM subtypes [13, 19]. Anti-synthetase autoantibodies may be detected before the onset of myopathy. However, EULAR/ACR classification criteria [12] used only the presentation of anti-Jo-1 autoantibodies. Some researchers point out that this is a limitation of the updated criteria [3]. Anti-Jo-1 is more common in adult patients with DM and correlated with more severe muscle involvement [19], but is rare in children. In none of the presented patients were anti-Jo-1 autoantibodies detected.
In contrast, anti-PL-7 autoantibodies were detected in all our patients. At the same time, literature sources indicate that in European cohorts, particularly in the UK cohort, anti-synthetase autoantibodies were detected only in 1.3% of patients with JIA and the majority of them were of Black ethnicity [21]. Moreover, anti-PL-7 autoantibodies were identified only in one patient (versus all patients in the present cohort). Anti-PL-7 and anti-PL-12 were associated with lung involvement [21], as was also demonstrated in another study [19]. Besides that, the combination of anti-Ro-52 with other anti-synthetase autoantibodies significantly increases the risk of ILD in patients with JDM; it is associated with chronic severe disease and poor prognosis [25–27]. None of 34 Indian patients with JDM had anti-synthetase autoantibodies, and overall the prevalence of myositis autoantibodies in this population was low [28]. On the other hand, the presence of anti-synthetase autoantibodies may indicate not only inflammatory myopathies, but also other connective tissue diseases [29].
Anti-Кu and anti-PM/Scl autoantibodies are associated with connective tissue disease overlap syndrome, including juvenile myositis overlapping with another autoimmune or connective tissue disease [26, 30], and mortality was highest in patients of this group [30]. Only one out of our patients had anti-PM/Scl and anti-Ku autoantibodies.
The inability to detect other myositis-specific autoantibodies and the small number of cases limit the full assessment of antibody status and correlation with the clinical phenotype. However, even the available data allow us to make certain conclusions and determine further follow-up of patients. Although there was no evidence of ILD in our patients at this stage of disease, lung status and function should be monitored to prevent further involvement [31, 32], as anti-synthetase autoantibodies have been identified in all children.
In 3 out of 4 patients we detected a combination of two or three myositis autoantibodies. Another study has also shown multiple autoantibodies in 15.3% of adults with DM [23]. For example, anti-Mi-2 autoantibodies that were associated with a favorable course in case 4 were combined with anti-PL-7 and anti-Ro52 autoantibodies, which increase the risk of ILD and correlated with severe course and poor prognosis [24, 27]. Therefore, the impact of a combination of different antibodies on the course of the disease requires further study.
The other issue concerns the influence of population and ethnicity on the presence of myositis-specific autoantibodies. We have already mentioned that there was a difference between the prevalence of anti-synthetase antibodies in the Indian population and in the UK [21, 28]. Besides that, research has demonstrated higher prevalence of antibodies and ILD in patient of Black or Hispanic descent [21, 33].
The patients were treated according to existing guidelines [34–36] and included the use of GCs with MTX in all cases. In two cases pulses with intravenous MP for 3 consecutive days followed by oral MP were used. In one case the treatment started with intravenous dexamethasone (2 mg/kg b.w./day) followed by oral MP and one patient received only oral MP in a dose of 2 mg/kg b.w./day. Glucocorticosteroid therapy had a positive effect in all cases, regardless of the different regime of administration. Other studies also demonstrated no significant difference in distinct GC regimens, although in severe cases it is recommended to use i.v. MP [36–38]. The use of MTX allows the disease to be controlled after weaning from GC therapy and allows the long-term use of high doses of GCs to be avoided, thus allowing their side effects to be reduced [39].
Conclusions
Juvenile dermatomyositis diagnosis may be difficult at the onset of the disease due to absence of signs of muscle involvement and/or mild specific dermatological features.
Detection of myositis autoantibodies helps to confirm the diagnosis of JDM as well as to determine the prognosis and algorithm for patients’ management. The presence of anti-synthetase syndrome in all the presented patients from one center, mainly due to the presence of anti-PL-7 autoantibodies, encourages further study with more patients and with determination of other myositis-specific autoantibodies to identify or refute certain regional features.