
Introduction
Idiopathic inflammatory myopathies (IIM) is a highly heterogenous group of connective tissue diseases. Several subtypes are distinguished, such as dermatomyositis (DM), polymyositis, antisynthetase syndrome, immune-mediated necrotizing myopathy and sporadic inclusion body myositis. It is now well recognized that the historical understanding of DM as inflammation of the muscles with the presence of cutaneous lesions is imprecise [1]. The clinical phenotype of DM extends from amyopathic to severe muscle weakness with a broad spectrum of extramuscular and extracutaneous organ involvement, including interstitial lung disease, vasculopathy or gastrointestinal complications [2]. The discovery of muscle-specific and muscle-associated antibodies led to the identification of serologically based clusters [3]. Myositis with anti-melanoma differentiation-associated gene 5 antibodies (anti-MDA5) is characterized by its distinct clinical presentation and poor outcome. However, even within this subtype, various clinical courses occur, leading to frequent diagnostic delay.
The study aims to review different courses of anti-MDA5-related myopathy, based on the cases of patients hospitalized in one academic centre.
Comprehensive data on the course of the disease in anti-MDA5-related cases were retrieved from the patients’ electronic medical records. The patients described in this manuscript have given written consent to publication of their case details and their photographs. To prepare the literature review, the PubMed database was thoroughly searched, using keywords such as “MDA5”, “MDA5 myositis”, “dermatomyositis”, and “vasculopathy”. Due to the rarity of the disease, the search criteria were not limited to certain years of publication. Original and review articles, as well as case reports, were considered. Articles published in English and Polish, relevant to the scope of this paper, were screened and selected papers were included (Fig. 1).
Case descriptions
Case 1
A 39-year-old female patient with asthma and pollen allergy presented initially with eyelid swelling and erythema triggered by exposure to sunlight. After a month she developed inflammation of the carpal joints, myalgia, muscle weakness, dyspnoea, xerostomia, and intermittent subfebrile states. She was evaluated in the outpatient clinic, because systemic connective tissue disease, mainly Sjögren disease, was suspected. Methylprednisolone was administered (20 mg/day), and the patient was referred to the rheumatology department.
On admission, laboratory tests revealed elevated alanine and aspartate aminotransferases (ALT, AST), mild thrombocytopenia, elevated erythrocyte sedimentation rate (ESR), and significant proteinuria (3,829.2 mg/24 h); while creatinine kinase, myoglobin, and C-reactive protein (CRP) were within the normal ranges. Antinuclear antibodies (ANA) speckled type were detected at a titre of 1 : 640. The standard immunoblot showed anti-Ro52 positivity, while myositis-specific autoantibodies immunoblot was not performed at that time. Peripheral consolidations of the lung parenchyma, mainly in the lower lobes, were found in chest computed tomography (CT). Electromyographic examination suggested a myopathic pattern and the Schirmer’s test showed normal tear secretion. Echocardiographic examination showed normal left and right ventricular systolic function (left ventricle ejection fraction [LVEF] = 62%). Based on the clinical image alongside proteinuria, systemic lupus erythematosus was suspected, although in the absence of lupus-specific antibodies and with normal complement concentrations. A kidney biopsy was performed, which revealed features typical of focal and segmental glomerulosclerosis (FSGS). Undifferentiated connective tissue disease was diagnosed. Methylprednisolone was slowly tapered, and chloroquine (250 mg/day) and nephroprotective treatment with angiotensin-converting enzyme inhibitor were added. Over time, novel symptoms appeared, such as ulceration of the fingers, redness and tenderness of the nail folds, erythematous lesions on the face and aphthous ulcers on the oral mucosa. Attempts of treatment with methotrexate (15 mg/week) then hydroxychloroquine (200 mg/day) were ineffective. Hand arthritis prevailed, and low-grade fever recurred. The patient complained of significant body mass loss (13 kg/year), alopecia and redness above the joints of the hand, consistent with Gottron’s sign. Dermatomyositis was suspected and the patient was referred to our department.
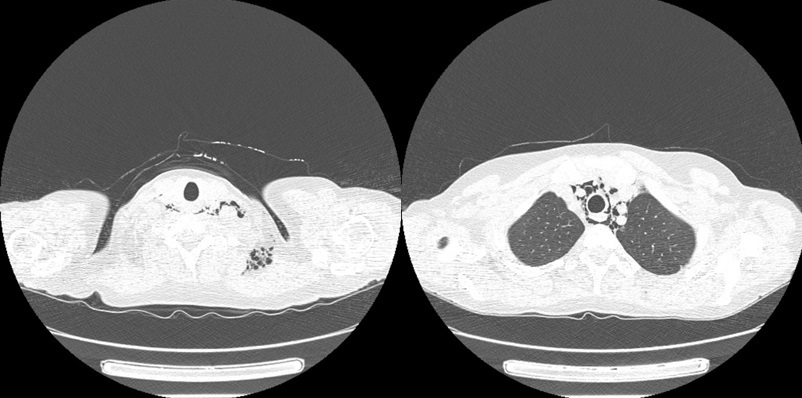
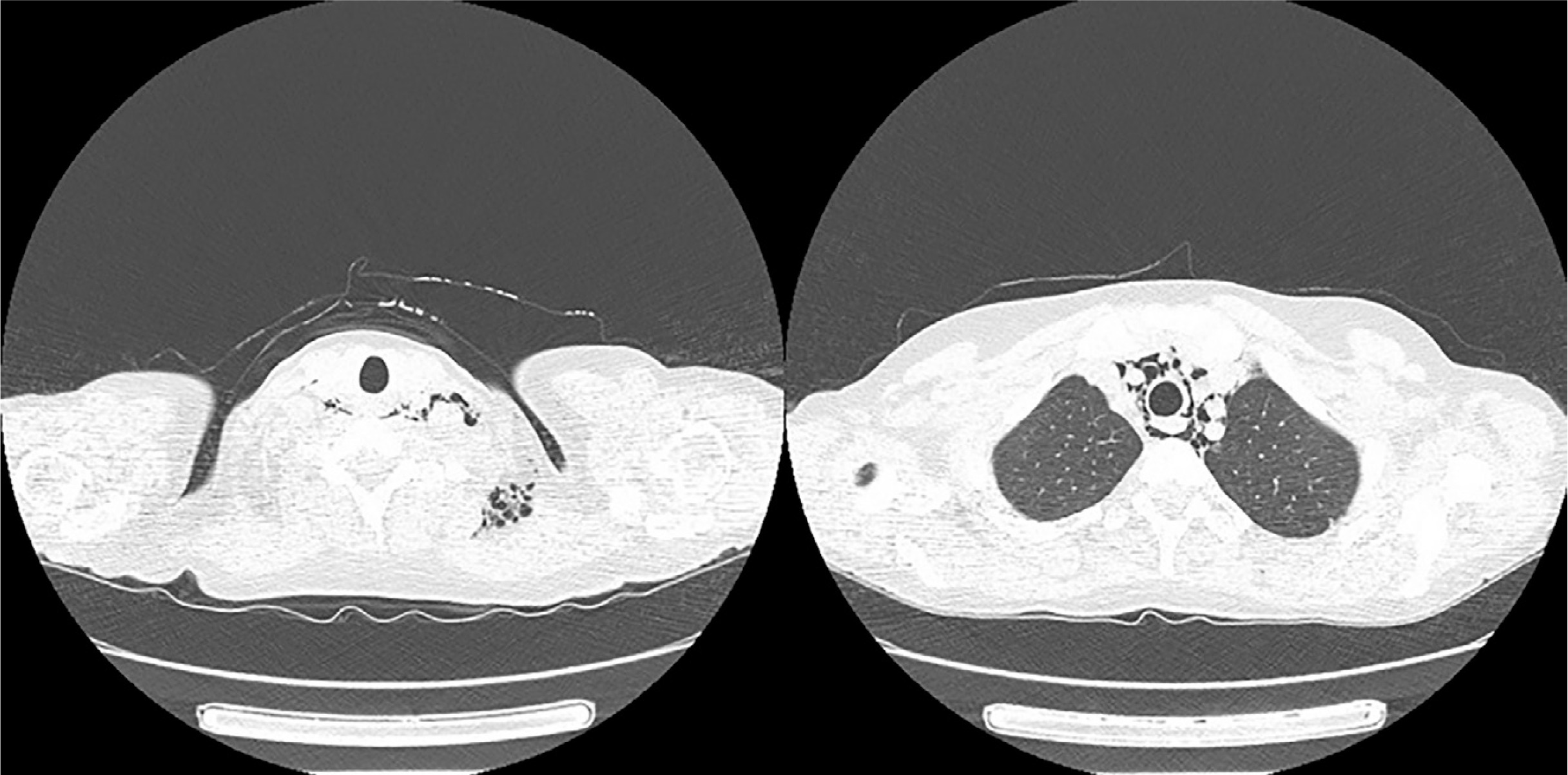
On admission she presented with slight proximal muscle weakness, subtle Gottron’s sign over the meta-carpophalangeal and proximal interphalangeal joints, palmar papules above the proximal and distal interphalangeal joints (inverse Gottron’s sign), nailfold erythema, subtle heliotrope rash, ulcerative lesions above both of the elbow joints and in the gluteal area, alopecia and isolated crackles at the bases of both lungs on auscultation. Laboratory investigations revealed hyperferritinaemia, elevated ESR and slightly increased level of AST and lactate dehydrogenase, with normal values of the remaining markers of muscle damage. Myositis-specific autoantibodies immunoblot (Euroimmun, Myositis Profile 3) revealed the strong positivity of anti-MDA5 and marked positivity of anti-Ro-52, with marginal values for anti-Jo1 and anti-PL7 antibodies. Chest CT showed interstitial lung disease with noticeable bilateral parenchymal opacities in lower lobes and “honeycombing” in the apices. Furthermore, chest CT revealed extensive yet asymptomatic pneumomediastinum and subcutaneous emphysema of the neck (Fig. 2). Bronchoscopy excluded damage to the bronchial tree, and an esophagram showed no leakage; therefore spontaneous pneumothorax was diagnosed. Computed tomography of the abdomen and pelvis showed no signs of malignancy. Echocardiographic examination revealed worsening of left and right ventricular systolic function (LVEF = 52%), compared to the examination performed a year earlier. Treatment with mycophenolate mofetil (2 g/day) was initiated and methylprednisolone (12 mg/day) was continued. Weight stabilized, but after two months of treatment myalgia and exacerbation of ulcerative skin lesions were observed. The patient was administered cyclophosphamide (CTX, 1,000 mg/month, 3 cycles), methylprednisolone (12 mg/day) and sildenafil (20 mg/day). Despite an initial modest improvement in the extent of the ulcerations, in the following months the disease progressed. Due to frequent infections of ulcerative lesions, the patient underwent topical antibiotic therapies. Cyclophosphamide was changed to cyclosporine (150 mg/day) with increased doses of methylprednisolone (16 mg/day) and sildenafil (25 mg/day). Skin ulcerations started to heal, and new lesions did not appear. Unfortunately, the ulcers continued to become infected, and special dressings and targeted antibiotic therapy were needed under supervision of a surgeon. After 6 months, disease remission was achieved. Due to the involvement of more than 20% of the lung parenchyma, nintedanib (150 mg twice a day) was added.
Fig. 2
Computed tomography scans with pneumomediastinum and subcutaneous emphysema detected in patient 1.
After almost a year of remission, disease relapse occurred. The patient reported general malaise, pain, swelling of the joints of the hands, and progressive muscle weakness. Skin lesions such as heliotrope rash, Gottron’s sign, palmar papules, and skin ulcers reoccurred. The patient was administered intravenous immunoglobulin (IVIG) therapy, with no clinical response. Based on the insufficiency of preceding therapies and promising results of Janus kinase (JAK) inhibitors in anti-MDA5 DM, it was decided to start off-label upadacitinib. Initially, gradual enhancement in muscle strength, reduction of arthritis and skin lesions was achieved. Nevertheless despite 3-month therapy, relapses of joint pain, skin lesions and fever were noted. Currently the patient is being treated with rituximab. Initial results seem to be promising as regards the constitutional symptoms and joint involvement. However, so far, skin ulcerations remain. Figure 3 presents the changes in ulcerative lesions over time.

Case 2
A 48-year-old female healthcare worker presented to the Emergency Department with dyspnoea and fever up to 39°C. The symptoms were accompanied by polyarthralgia, muscle weakness, general malaise and morning productive cough. Anamnesis revealed a history of sinus tachycardia, gastroesophageal reflux disease, increased levels of anti-thyroid peroxide and antithyroglobulin antibodies without clinically active thyroid disorder. Lung CT scan revealed diffuse infiltrates in the subpleural space in the bases of both lungs, thickening of interlobular septa and ground glass opacities, indicative for interstitial lung disease (ILD) in the phenotype of organizing pneumonia (OP). Swab test for COVID-19, flu and respiratory syncytial virus were negative.
Upon admission to the pneumonology department, the patient presented with heliotropic erythema accompanied by desquamating forehead lesions, Gottron’s papules above the small joints of the hands, nailfold erythema, telangiectasia, and swelling and tenderness of carpal and intercarpal joints. On auscultation, crackles in the bases of both lungs were detected. Laboratory investigations revealed elevated levels of ALT, AST and CK. Inflammatory markers such as CRP and ESR were subtly elevated, anti-cyclic citrullinated peptide antibodies likewise. Rheumatoid factor (RF) level was within the normal range. Antinuclear antibodies were positive, at a titre of 1 : 640, with 1 : 320 speckled type and 1 : 160 cytoplasmatic type. Myositis-specific antibodies immunoblot revealed strong positivity of anti-MDA5, while in ANA 3 profile, anti-Ro-52 and anti-DFS-70 antibodies were detected. There were no obstructive or restrictive abnormalities in spirometry. Plethysmography showed correct total lung capacity, with moderate diffusion disturbance in diffusion lung capacity for carbon dioxide (DLCO – 68%). The patient achieved 410 metres in the 6-Minute Walk Test without desaturation. A diagnosis of anti-MDA5-related DM with ILD-OP was proposed, based on the opinion of a multidisciplinary team (pneumonologists, rheumatologists, radiologists), and the patient was referred to the Rheumatology Department.
Upon admission to the Rheumatology Department (Fig. 4), the patient complained of weakness of the muscles of the pelvic and shoulder girdle, which led to difficulties in standing up from the recumbent position, stair climbing and arm raising. The symptoms were accompanied by exertional dyspnoea, fever with night sweating, dysphagia, arrhythmia, fatigue and progressive weight loss (5 kg in 2 months). Gastroscopy was conducted yet excluded dysplasia. Echocardiography revealed preserved systolic contractility of the left ventricle (left ventricular ejection fraction 66%). Heart magnetic resonance imaging (MRI) detected fibrotic lesions in the myocardium of the left ventricle, without features distinctive for ischemic aetiology. Capillaroscopy examination detected numerous microhaemorrhages and blood vessels in the breakdown phase. The treatment was initiated with intravenous pulses of methylprednisolone (500 mg/day for 3 days) with subsequent slowly tapered oral prednisone in combination with cyclosporine in the dose of 150 mg/day. The patient reported significant improvement of muscle strength, and resolution of fever and night sweating. However, pain of the fingertips, finger spasms, itching sensation of the eyelids and Raynaud’s phenomenon occurred. Due to only partial remission, IVIG were added to the ongoing treatment, which ultimately resulted in improvement of muscle strength and additional symptoms. The administered treatment yielded remission of all cutaneous lesions, except slight erythema on the palmar hand surface. The CT scan was retaken at 3 and 5 months after the disease onset, revealing stable interstitial lesions.
Case 3
A 54-year-old female nurse with a history of hypothyroidism, pulmonary and mesenteric thromboembolisms related to hypercoagulability with heterozygotic mutation of V Leiden factor complained of fatigue, loss of appetite, weakness of the proximal muscles of the upper and lower extremities, and pain and swelling of carpal and intercarpal joints. Due to the muscle weakness, the patient had difficulties with standing up from sedentary and recumbent positions. Significant weight loss in a short time (20 kg in 3 months), arrhythmia and occasional dry cough without dyspnoea were also observed. The diagnosis of rheumatoid arthritis was made (positive RF, 10 painful and 8 swollen joints, Visual Analogue Scale [VAS] 8/10), and treatment with hydroxychloroquine (200 mg/day) and methylprednisolone (4 mg/day) was initiated. After 2 months, methotrexate in a dose of 15 mg weekly was added. Despite the treatment, the patient’s condition deteriorated. She complained of progressive muscle weakness and dysphagia. Moreover, heliotrope rash, Gottron’s sign and inverse Gottron’s sign appeared. Due to ineffectiveness of the proposed therapies, the patient decided to discontinue the treatment.
In the meantime, painful ulcers on the tips of each finger of the right hand and above the elbow joints emerged, as well as distal necrotic lesions at the tip of the left third finger. The ulcers became infected and were treated with antibiotics. The patient was re-evaluated in the rheumatology outpatient clinic and anti-MDA5-related DM was suspected. Laboratory examinations revealed lymphopenia, neutrophilia, mild thrombocytopenia, elevated inflammatory markers and hypercholesterolemia. Non-contrast chest CT revealed slight bilateral bronchiectasis and reticular fibrous lesions covering < 20% of the lung parenchyma resembling a honeycomb image. Moreover, imaging exposed nodular goitre with right-side trachea dislocation, and heart enlargement with pericardial fluid (13 mm). The flow-volume spirometry test excluded upper respiratory tract obturation and confirmed restrictive disorder (forced vital capacity 74%). Diffusion lung capacity for carbon dioxide was moderately decreased (57%). Antinuclear antibodies were detected, at a titre of 1 : 320 speckled type, 1 : 160 cytoplasmatic type and 1 : 160 filament type. Myositis-specific antibodies immunoblot revealed presence of anti-MDA5 antibodies. Methylprednisolone (24 mg/day) in combination with cyclosporine (125 mg/day) was administered.
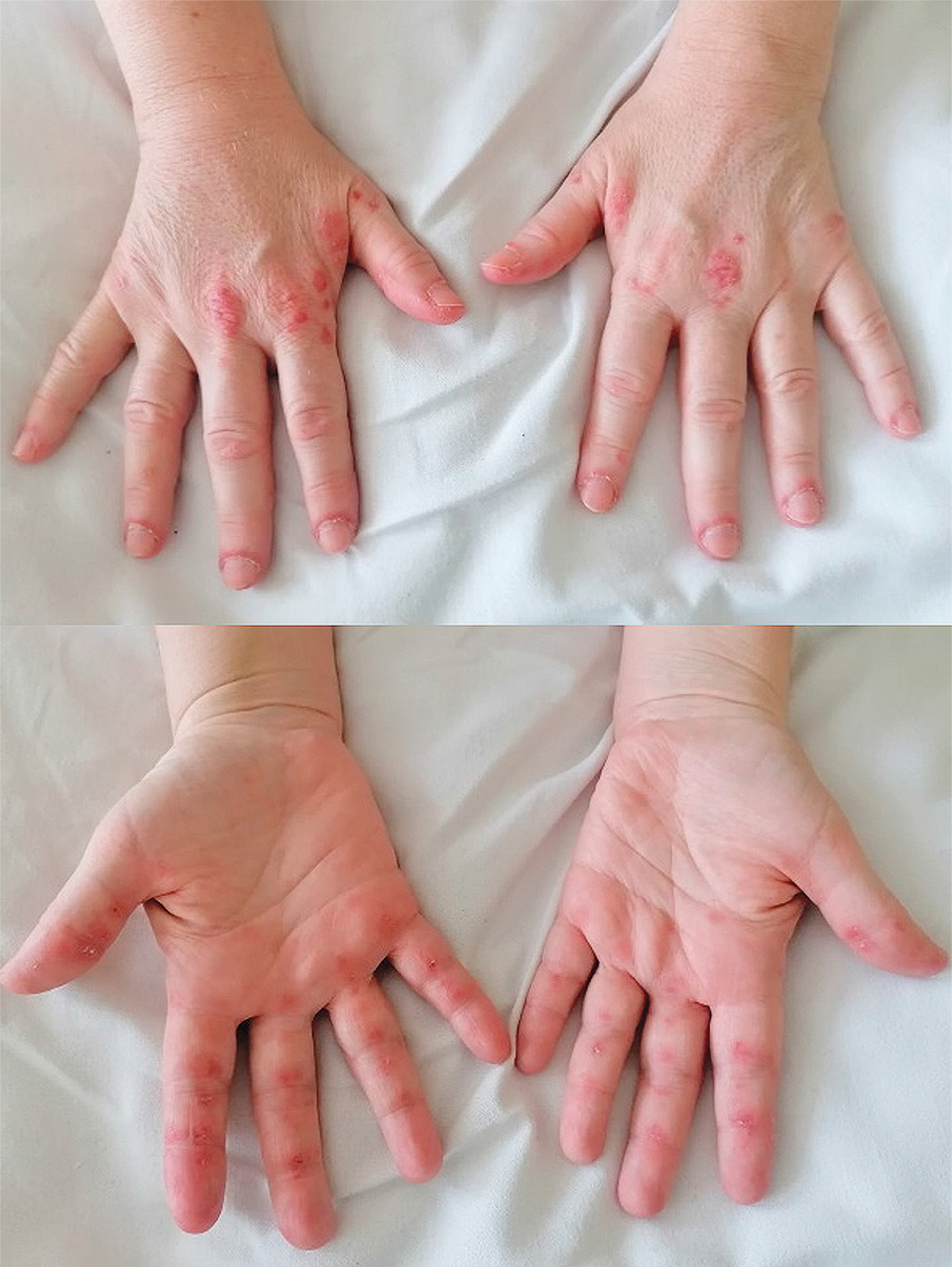
Significant improvement was observed in muscle strength and joint involvement. However, progression of skin ulcerations was observed. Crackles in both lungs were present on auscultation. Therapy with intravenous pulses of CTX (1,000 mg monthly) was initiated. The daily dose of cyclosporine was increased to 150 mg and the methylprednisolone dose was slowly tapered. Gradually, a reduction in skin ulcers and joint pain and an increase in muscle strength were observed. The patient also underwent botulin toxin injection into the right hand and fingers, leading to a reduction of ulcerations, relief of pain and increased joint mobility. After 3 months of CTX and 4 months of cyclosporine therapy, the patient reported significant improvement of muscle strength, resolution of joint and muscle pain, and alleviation of distal necrotic lesions of fingers. Figure 5 presents the evolution of skin lesions during the disease.
Fig. 5
Evolution of cutaneous lesions in patient 3: A) Gottron’s sign and inverse Gottron’s sign at the beginning of the disease; B) progression of the ulcerative lesions, necrosis of the distal phalange of the 4th right finger; C) improvement after pharmacotherapy and botulin toxin A injection.

Table I summarizes and compares the main laboratory results at IIM diagnosis in the three presented patients. As the data were analysed retrospectively, some results as well as core set measure assessment are missing, which we consider to be a limitation of the study.
Table I
Laboratory results of the patients at the time of IIM diagnosis
Discussion
Anti-melanoma differentiation-associated gene 5 anti-bodies are a relatively novel serological marker, first described in 2005 [4]. Significant geographical variation in the frequency of incidence is observed. In European cohorts, anti-MDA5-related DM is a rare entity. According to the EuroMyositis database, anti-MDA5 antibodies were detected in 1.3% of all IIM patients [5]. This is in line with our centre’s experience. In contrast, in Asian countries anti-MDA5 DM is far more prevalent, with predominance in South-East Asia [6]. Regardless of ethnicity, women constitute the overwhelming majority of patients [6]. All our three patients were Caucasian females.
Melanoma differentiation-associated gene 5 is gaining recognition as the critical agent of the interferon pathway. Encoded by the IFN-inducible gene 1, it serves as a cytosolic sensor for double stranded RNA (dsRNA), which derives from viral pathogens but also endogenous mitochondria. Viruses capable of MDA5 activation include picornavirus, flavivirus, coronavirus and herpesvirus families [7, 8]. Upon stimulation with dsRNA, MDA5 initiates interferon response with transcription of interferon-dependent genes [2, 9]. Considering the crucial role of MDA5 in the viral-induced response, viral infections seem like plausible triggers of anti-MDA5 syndrome. This hypothesis is confirmed by the seasonality of disease onset with a peak in late winter and spring [10, 11]. In our patients, symptoms of anti-MDA5-related myositis started during different seasons of the year (case 1 – August, case 2 – January, case 3 – April). Several similarities were noted between clinical presentations of COVID-19 and dermatomyositis with presence of anti-MDA5 autoantibodies [12, 13]. Cases of new-onset anti-MDA5-related DM shortly after SARS-CoV-2 infections have been reported [14, 15]. However, in our patients no such association was found.
Usually, DM with anti-MDA5 antibodies is clinically amyopathic. Muscle biopsies reveal no or only subtle features typical for DM such as perifascicular fibre atrophy, tubuloreticular formation, capillary loss or inflammatory infiltration. However, some unique features are observed, for example nitric oxide synthase 2-positive muscle fibres [16]. According to Betterigde et al. [5], DM phenotype with presence of anti-MDA5 was negatively associated with raised CK. Our patients experienced muscle weakness and/or myalgia, although with normal or insignificantly elevated muscle enzymes. Due to frequent extramuscular involvement, the term “anti-MDA5+ syndrome” instead of anti-MDA5-related dermatomyositis was proposed [17]. Joint involvement usually affects small joints of the hands and wrists, which may resemble rheumatoid arthritis or psoriatic arthritis [18, 19]. All our patients presented with polyarthritis, which responded well to therapy. In contrast to muscle involvement, in anti-MDA5-related myopathy cutaneous lesions are typical, including both symptoms shared by all DM types, such as heliotrope rash, V sign, shawl sign, Gottron’s sign or mechanic’s hands, and lesions specific for this subtype, such as inverse Gottron sign (palmar papules, especially over metacarpophalangeal and interphalangeal joints, frequently painful, hyperkeratotic or ulcerative), skin ulceration, alopecia or auricular skin lesions [6, 20]. Skin lesions were present in all our patients – both those typical for DM and anti-MDA5 specific. Tender gums and oral ulcers, observed in this subtype of IIM, may mimic systemic lupus erythematosus [20]. This was also true for one of our patients (case 1). In anti-MDA5-related myositis, ulcerations are typically deep and painful, localized over the Gottron’s papules, elbow and knee extensor surfaces, fingertips and nailfolds. In some patients, calcinosis cutis may co-occur [2]. These symptoms reflect ongoing vasculopathy, inflammatory infiltration of the vascular wall and intravascular thrombi [20]. Wounds are difficult to heal and may become infected, leading to systemic infection or even amputation [21, 22]. We also observed this complication in two of our patients (cases 1 and 3). The critical complication of anti-MDA5-related myopathy is ILD, which is often severe and rapidly progressing. Depending on the studies, ILD was observed in 42–100% of the patients and is more prevalent in Asian populations as compared to European and US cohorts [23]. Patients with anti-MDA5 antibodies were found to have over 20-fold higher risk of developing rapidly progressive ILD (RP-ILD) as compared to anti-MDA5 negative patients [24]. Rapidly progressive ILD remains one of the leading causes of death, significantly worsening the prognosis, especially at the first stage of the disease [6, 25]. Different patterns of ILD are observed, but the most common one is the “unclassified” consolidation-based pattern [26]. Interstitial lung disease may be complicated by pneumomediastinum, which occurred in one of our patients. It is considered a negative prognostic factor associated with high mortality [27, 28]. All our patients developed ILD. In another patient organizing pneumonia preceded symptoms of anti-MDA5-related myopathy. Rapid progression of ILD was not observed, probably as aggressive immunosuppression was promptly implemented. Finally, anti-MDA5-related phenotype is marked by inflammation, which is frequently reflected by constitutional symptoms such as fever, general malaise or fatigue. Significantly elevated ferritin is considered to be a risk factor for ILD and poor prognosis [6]. Notably, two of the three described patients had fever, and in one, significant weight loss was noted. In all of the patients, ferritin was elevated. Renal involvement is unusual for IIM, including anti-MDA5-related type. A single case of a patient with nephrotic syndrome and biopsy-proven thrombotic microangiopathic glomerular lesions in the course of anti-MDA5 myositis was reported [29]. Interestingly, one of our patients (case 1) presented with persistent proteinuria. Renal biopsy demonstrated FSGS, which to our knowledge has never been documented in an anti-MDA5-related patient.
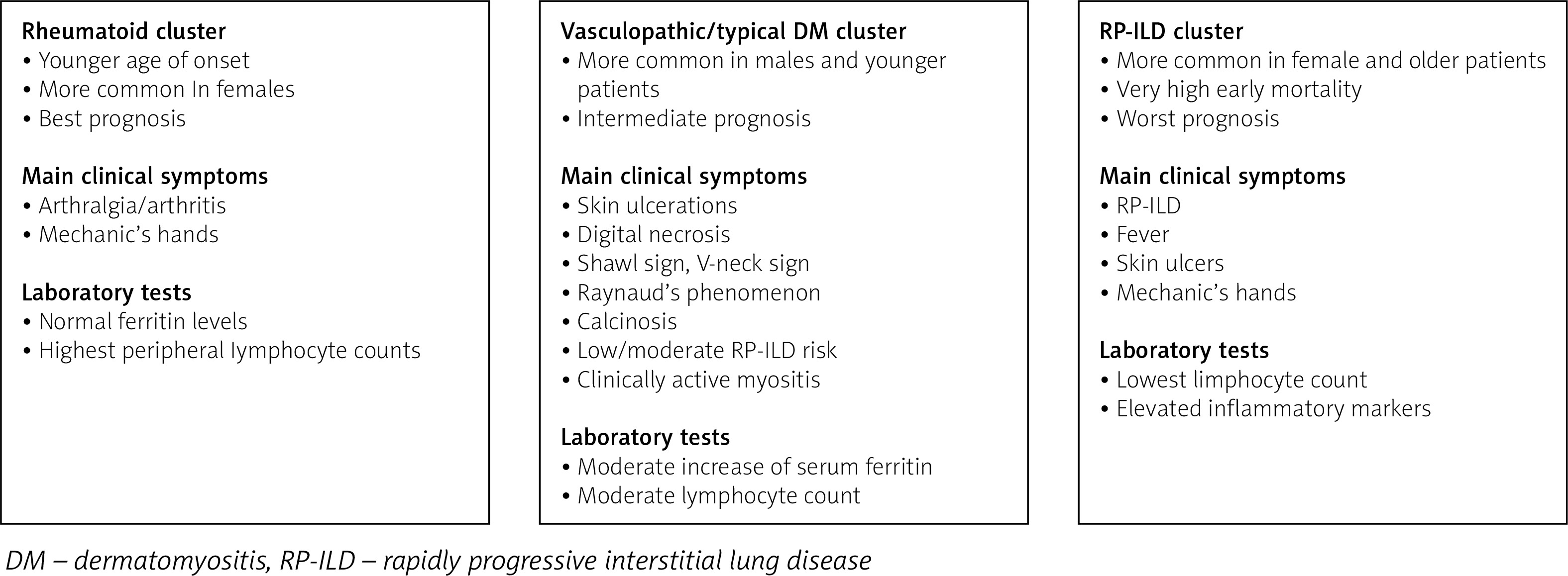
Despite quite a consistent and specific spectrum of symptoms, courses of anti-MDA5-related myopathy differ among individual patients. Several studies have identified phenotypes of anti-MDA5-related myopathy. In the multicentre study by Allenbach et al. [17], 3 clusters were distinguished: the first, associated with RP-ILD, which included patients (mostly women) with severe ILD, frequent mechanic’s hands, poor prognosis and very high early mortality; cluster 2 was a rheumatoid cluster, characterized by frequent arthralgia/arthritis, higher prevalence in women and significantly better prognosis as compared to other clusters; cluster 3 – vasculopathic – included mostly men with severe skin vasculopathy, Raynaud’s phenomenon, cutaneous ulcers, clinically active myositis and a tendency to digital necrosis and calcinosis, who were found to have an intermediate prognosis with relatively low early mortality. Similar observations were noted in a smaller group of patients by Labrador-Horillo et al. [30], who also distinguished three phenotypes: patients with amyopathic DM and RP-ILD, characterized by high mortality and poor prognosis despite treatment; patients with a phenotype resembling antisynthetase syndrome; and a subgroup of patients with amyopathic DM but prominent skin involvement. According to this clustering, our patients could be classified into the rheumatoid cluster (case 2) and vasculopathic cluster (cases 1 and 3). The proposed clustering of patients with anti-MDA5-related DM is presented in Figure 6. Different phenotypes were identified by Yang et al. [31], including patients with classic dermatomyositis, with muscle weakness, typical skin lesions and good prognosis; a subgroup with predominant arthritis, mechanic’s hands and intermediate prognosis; and a subgroup with poor prognosis manifesting with RP-ILD, fever, and elevated inflammatory markers. Xu et al. [32] divided patients with anti-MDA5-related myopathy depending on the risk of RP-ILD into patients with mild, moderate and high risk. In patients with high risk of RP-ILD, anti-Ro52 antibodies were more prevalent. Interestingly, anti-Ro52 antibodies were detected in our patient 1, who developed ILD with pneumomediastinum.
Treatment of anti-MDA5 syndrome is challenging, and many patients remain refractory to even aggressive therapy. Little evidence is supported by randomized trials, and the consensus comes mostly from observational studies or case reports. Recommendations for the treatment of RP-ILD in the course of anti-MDA5-related myositis support the combination of high-dose glucocorticosteroids (GCs) with calcineurin inhibitors (CNI) and with the additive use of CTX in more severe cases [33]. This triple therapy (GCs + CNI + CTX), also known as the “Japan protocol”, has well-documented efficiency and significantly improved patients’ survival. Notably, as most patients were diagnosed and treated at the early stage of the disease, the effects of treatment in long-lasting disease may not be so promising [34]. Moreover, the proposed treatment regimen was associated with a high risk of opportunistic infections [35]. According to a randomized controlled trial, tacrolimus may be a more effective option than cyclosporine, as it improved the results of pulmonary functional tests to a greater extent, although no differences in the overall survival were noted [36]. In recent years JAK inhibitors have emerged as a promising therapeutic option in ani-MDA5-related myositis. A significant improvement in the survival and a good safety profile were demonstrated in a group of patients with early anti-MDA5-related ILD treated with a combination of tofacitinib and GCs [37]. Several cases of patients who responded to tofacitinib despite the ineffectiveness of triple therapy have also been described [38, 39]. According to the literature, rituximab should also be considered in patients with MDA5 syndrome, as it was found to improve both cutaneous lesions and pulmonary function. Both conventional dosage and low-dose therapy proved to be effective, with a lower risk of infection for the latter regimen [40]. Patients with anti-MDA5 myopathy also benefited from adjunct IVIG, which led to statistically significantly lower mortality and sustained remission [41]. According to the recommendations, IVIG, as well as plasmapheresis or polymyxin B hemoperfusion, should be considered as a rescue therapy [33]. Extracorporeal membrane oxidation could serve as a bridge therapy in life-threatening conditions or before lung transplantation [12]. Attempts are being made to treat vasculopathy with calcium channel blockers, phosphodiesterase inhibitors, pentoxifylline or even hyperbaric oxygen, but so far limited data are available [12, 42]. Botulinum toxin injections have gained recognition as a method of treating difficult-to-heal wounds, including ulcerations caused by vasculopathy, e.g. in systemic sclerosis. Injections of botulinum toxin type A were effective in 81% of patients with ulcers due to Raynaud’s phenomenon [43]. Apart from immunosuppressive therapy, adding an antifibrotic drug such as nintedanib or pirfenidone is justified in patients with IIM-ILD. Several studies have demonstrated reduced mortality or lower risk of rapid progression in patients receiving additively antifibrotic drugs [44–46]. One of our patients (case 1) is being treated with nintedanib, while in the other patients stabilization of pulmonary lesions does not justify the addition of this drug for now.
Conclusions
Anti-MDA5-related myositis is characterized by a unique clinical presentation, which significantly differs from other myopathies. Due to the unusual phenotype and frequent amyopathic presentation, establishing the correct diagnosis remains challenging. Low awareness of clinicians about this rare subtype contributes to the significant diagnostic delay, hindering effective treatment. Within anti-MDA5 myositis, three main phenotypes may be distinguished, enabling prediction of the prognosis and risk of organ involvement in affected patients. Various treatment regimens are being developed, yet there is a consensus that aggressive immunosuppression should be implemented at the very early stage of the disease to prevent organ-threatening complications.