
Introduction
Hyperuricemia is a common issue in daily clinical practice, estimated to occur in approximately 8.9% to 24.4% of the general population [1, 2]. Although its definitions may differ slightly depending on laboratory norms and test methods, hyperuricemia is usually defined as serum uric acid (UA) concentration higher than 7 mg/dl [3].
Age- and sex-specific norms for serum UA are generally accepted as 3.5 to 7.0 mg/dl (208–416 µmol/l) in men and postmenopausal women, and 2.6–5.7 mg/dl (155–339 µmol/l) in premenopausal women [4]. These values have been determined based on UA solubility capacity.
Above the threshold of 6.8 mg/dl the risk of serum UA crystallization increases significantly [1]. The observed differences between sexes have been attributed to estrogen’s stimulating effect on UA renal excretion [5].
Hyperuricemia can be classified as symptomatic (when accompanied by symptoms of gout, urolithiasis or acute urate nephropathy), or asymptomatic (defined as increased serum UA level without any of the symptoms mentioned above) [3].
Recently, there has been a growing interest in the role of UA in a variety of health disorders. Although a majority of the asymptomatic patients never develop changes that could be attributed to urate crystals deposition, it has been found that elevated serum UA levels may cause silent tissue damage and increase the risk of several diseases such as hypertension, dyslipidemia, obesity, metabolic syndrome, type 2 diabetes, cardiovascular disease and chronic renal disease (CKD) [1, 4].
Furthermore, hyperuricemia is a prognostic marker of mortality in patients with stable coronary artery disease treated with percutaneous coronary interventions [6], patients with chronic obstructive pulmonary disease [6] and terminally ill cancer patients [7].
In critically ill patients in intensive care units, hyperuricemia is an early marker of the severity of sepsis, as well as a predictor of acute kidney injury (AKI), acute respiratory distress syndrome, need for mechanical ventilation use and mortality [8, 9].
Whether asymptomatic hyperuricemia should be treated and in which patients, what the UA threshold for starting urate-lowering therapy (ULT) should be, whether routine testing for UA serum level an appropriate approach, and, finally, whether UA is a relevant prognostic tool in clinical screening are questions relevant to clinicians of diverse specialties.
The aim of this study is to provide answers to these questions based on a review of the current literature.
Uric acid origin, metabolism and role in the human body
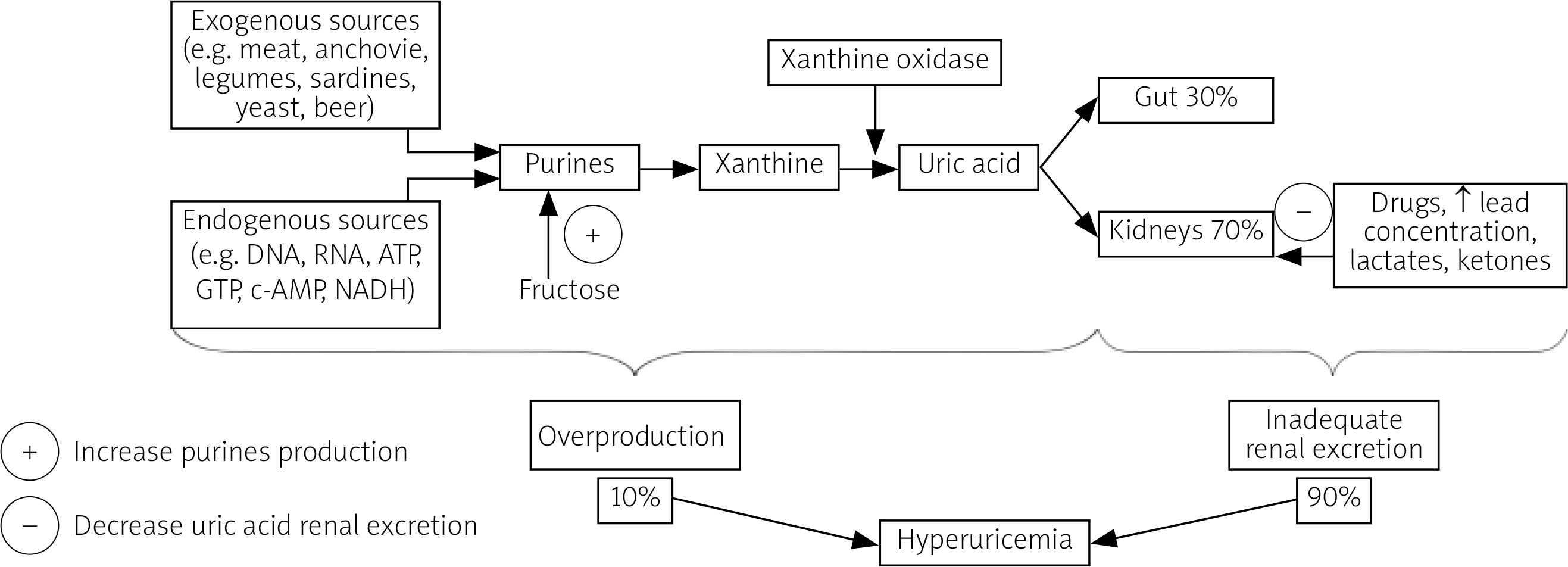
Uric acid is a weak acid generated as the final product of the metabolism of purines, which form important biologic particles such as the DNA, RNA, ATP, GTP, c-AMP and NADH [4].
The vast majority of serum UA comes from endogenous sources (e.g. nucleic acid breakdown and de novo purine biosynthesis). The daily endogenous purine production is estimated to amount to about 500–600 mg, while intake of exogenous purines with diet is approximately 100–200 mg per day [10].
Dietary products rich in purines include organ meats (especially the liver and the kidneys), anchovies, legumes, sardines, yeast and beer [3, 4].
Although products rich in fructose are not a direct source of purines, their consumption can increase serum UA production by intensifying adenosine triphosphate (ATP) degradation [11].
The crucial enzyme catalyzing purines conversion to UA in humans and a target of several drugs is xanthine oxidase. Its highest concentration is observed in the liver, which is the main organ of UA production. However, xanthine oxidase is also present in other organs such as the intestines, the kidneys, the lungs, the heart, the brain, muscles and vessels [1, 4].
Interestingly, UA catabolism is different in human beings compared to other mammalian species. In humans, UA is the final product of purine metabolism, while the majority of other mammals produce uricase that breaks down UA into better soluble allantoin, easily excreted with urine. One hypothesis assumes that the uricase-coding gene has been lost in the course of evolution in order to maintain a higher blood pressure, necessary to uphold the upright body posture [12].
According to some authors, the loss of uricase was also helpful in the process of obtaining fat from the transformation of fructose contained in fruit, the main nutrient of human ancestors [13].
Yet another hypothesis based on the UA anti-oxidant potential assumes that UA as a strong anti-oxidant could have neuroprotective properties and thus play an important role in the development of human intelligence. By scavenging free radicals, UA could also extend lifespan and reduce cancer risk in humans and higher primates [12].
These hypotheses are supported by results from a recent study performed by Song et al. [14] showing potential protective and anti-oxidative UA effects on erythrocytes. In their study performed on 10,759 participants, a positive association between UA level and erythrocyte count as well as hemoglobin concentration was observed.
Furthermore, results from a study on the anti-oxidant role of UA performed on 356 Australian women revealed that higher UA level was related to higher bone mineral density. Authors suggested that UA could be a protective factor against osteoporosis [15].
Despite many hypotheses explaining the function of UA, its exact role in the human body remains unclear. Paradoxically, UA shows both antioxidant and pro-oxidant effects in a microenvironment-dependent manner. It is presumed to exert anti-oxidant activity mainly extracellularly (primarily protecting erythrocyte lipid membranes from oxidative damage and protecting neurons from demyelination by blocking peroxynitrite), while acting like a pro-oxidant inside the cell [4].
The pro-oxidative activities of UA may result in endothelial injury leading to hypertension and cardiovascular disorders [16], disruption of the oxidative balance inside the adipose tissue resulting in a higher risk of metabolic syndrome and insulin resistance [17], renal ischemia due to decreased nitric oxide levels and increased local vasoconstriction [18].
The kidneys play the key role in maintaining UA metabolic balance. Hyperuricemia is caused by inadequate renal excretion in about 90% of instances and excessive production in only 10% of cases. Kidneys eliminate about 2/3 of UA produced daily and the remaining 1/3 is removed through the digestive tract. Approximately 90% of the filtered UA load is reabsorbed by the renal proximal tubules, presumably due to its important physiological functions [10].
Renal re-absorption of UA involves several transporters such as urate transporter 1 (URAT1) and glucose transporter 9 (GLUT9), whose function may be influenced by a number of factors, e.g. genes, drugs, increased serum lead, lactate or ketone concentrations [1, 4]. The imbalance between UA production and elimination, dependent mainly on renal excretion, results in hyperuricemia (Fig. 1).
Gout and urate-lowering therapy
According to epidemiological studies performed on the United States population, gout affects about 1/3 of patients with hyperuricemia. However, gout arthropathy is estimated to be correctly diagnosed only in about 2/3 of affected subjects and properly treated in about half of diagnosed patients [19].
Pathophysiology of gout includes UA crystallization and subsequent mechanical and chemical irritation of articular tissues, resulting in inflammation of joints and soft tissues, as well as generation of tophi made of UA deposits.
Although algorithms concerning treatment of acute gout are generally agreed upon, some controversy surrounds the subject of chronic gout management, secondary gout prevention and use of UA-lowering therapy in asymptomatic patients.
Recommendations for management of hyperuricemia in patients diagnosed with gout are outlined in guidelines issued by the American College of Rheumatology (ACR) in 2012 [20], updated in 2020 [21], the “3E” Initiative (the Evidence, Expertise, Exchange Initiative) guidelines published in 2014 [22] and the European League Against Rheumatism (EULAR) recommendations issued in 2016 [23]. Similarities and disparities between them are presented in Tables I and II, respectively.
Table I
ACR (2012, 2020), 3E Initiative (2014) and EULAR (2016) recommendations for management of hyperuricemia in patients with acute and chronic gout – similarities [25–28]
Table II
ACR (2020), 3E Initiative (2014) and EULAR (2016) recommendations for management of hyperuricemia in patients with acute and chronic gout – differences [25–28]
In patients previously diagnosed with gout, sudden high purine intake with the diet increases the risk of gout recurrence almost five times [24]. Potentially beneficial dietary changes in secondary gout prevention include decreased intake of red meat, alcohol (especially beer) and sugary beverages, as well as increased coffee consumption [25].
Vitamin C supplementation is no longer recommended due to scarcity of data on its utility in gout prevention [21]. In overweight or obese patients, weight loss is recommended [21].
However, it should occur gradually in order to avoid a ketonemia-induced acute gout crisis. Although diet restriction seems less effective than ULT in lowering UA levels, dietary interventions in addition to ULT help achieve target UA levels.
As for pharmacological interventions, recommended first-line drugs are xanthine oxidase inhibitors (such as allopurinol and febuxostat). These drugs block transformation of UA precursors to UA. The most commonly used drug is allopurinol, a purine-selective xanthine oxidase inhibitor. The usual starting dose is ≤ 100 mg/day.
Authors of the American guidelines underline that in selected populations, i.e. of Southeast Asian or African American descent, prior to therapy with allopurinol, testing for the HLA-B*5801 allele should be considered due to increased risk of allopurinol-induced hypersensitivity reactions including a potentially life-threatening drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome [20, 21].
Other adverse reactions to allopurinol include rash, pruritus, cytopenia, diarrhea, toxic epidermolysis and Stevens-Johnson syndrome. The use of allopurinol in standard doses may also cause renal failure progression in patients with preexisting renal insufficiency. Renal clearance of oxypurinol, the major metabolite of allopurinol, is directly proportional to the renal creatinine clearance. Therefore, in patients with renal insufficiency, allopurinol should be used in reduced doses (e.g. starting dose 50 mg/day) in order to prevent life-threatening toxicity [26].
In the case of allopurinol’s inefficiency or intolerance, febuxostat, a non-purine selective inhibitor of xanthine oxidase, may be prescribed. The two xanthine oxidase inhibitors should not be used at the same time. The recommended starting dose of febuxostat is 40–80 mg/day.
If the target UA level determined by the UA crystallization threshold of about 6.8 mg/dl is not achieved after two weeks, the dose of febuxostat may be increased by 40 mg/day. Febuxostat is metabolized in the liver and the most common adverse reaction to this drug is a mild transaminases elevation [27].
In order to achieve the therapy target defined as complete resolution of residual symptoms including tophi and UA below target level, a uricosuric agent may be added to a xanthine oxidase inhibitor or used as monotherapy. Uricosurics increase UA renal excretion by inhibiting URAT1 in renal tubules.
According to the latest ACR guidelines, checking UA excretion prior to starting treatment with uricosurics and urine alkalization are no longer recommended. During the uricosuric therapy, an increased fluid intake may reduce the risk of urolithiasis [21, 28]. Uricosurics should be avoided in patients with renal calculi and/or stage III–V CKD [21]. Probenecid is probably the most commonly used uricosuric. The American College of Rheumatology recommends 500 mg once or twice daily and dose titration in order to achieve the therapy target [21].
Benzbromarone has been withdrawn from the USA and most European countries due to severe hepatotoxicity,but is still restrictively used in Asia. Between 2015 and 2016 the novel uricosuric drug lesinurad was approved by both the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) as second-line therapy.
This selective UA reabsorption inhibitor blocks both URAT1 and the organic anion transporter 4 (OAT4) in the renal proximal tubule. In comparison to probenecid, lesinurad is more potent and effective in moderate renal insufficiency.
Its administration at a dose of 200 mg/day in addition to either allopurinol or febuxostat allows the therapy target to be achieved in the case of xanthine oxidase inhibitor inefficiency [29, 30]. Higher doses of lesinurad such as 400 mg/day are associated with an increase in creatinine levels and therefore not recommended [31].
Some drugs used in therapy of hyperlipidemia and hypertension such as statins, fibrates and losartan increase UA excretion, and may potentiate the uricosuric effect [6, 8].
On the other hand, loop diuretics and thiazides elevate serum UA levels in gout patients [32]. These diuretics decrease UA excretion by competing with UA for cell membrane transporters in renal proximal tubules. According to the newest American guidelines, hydrochlorothiazide should be generally avoided in gout patients, if possible [21].
Another new drug which may be used as ULT in gout patients is arhalofenate, a peroxisome proliferator-activated receptor-gamma (PPAR-γ) partial agonist. By inhibiting urate transporter 1 (URAT1), organic anion transporter 4 (OAT4), and organic anion transporter 10 (OAT10), as well as IL-1β expression, it shows both UA-lowering and anti-inflammatory effects.
Patients on ULT usually receive anti-inflammatory prophylaxis in order to minimize the risk of gout attacks precipitated by the ULT itself. Arhalofenate at a dose of 800 mg/d is superior to allopurinol in reducing risk of gouty flares [33]. As for its anti-inflammatory effect, arhalofenate is weaker than conventional nonsteroidal anti-inflammatory drugs (NSAIDs) [34].
Uricases such as rasburicase or pegloticase (the PEGylated form) are second-line drugs. Their use is limited to severe and recurrent gout, due to parenteral administration and risk of infusion reactions. Uricases should be administered only as monotherapy, in the case of inefficiency of first-line drugs. Their prolonged use may be associated with production of anti-drug antibodies and thus decrease their efficacy.
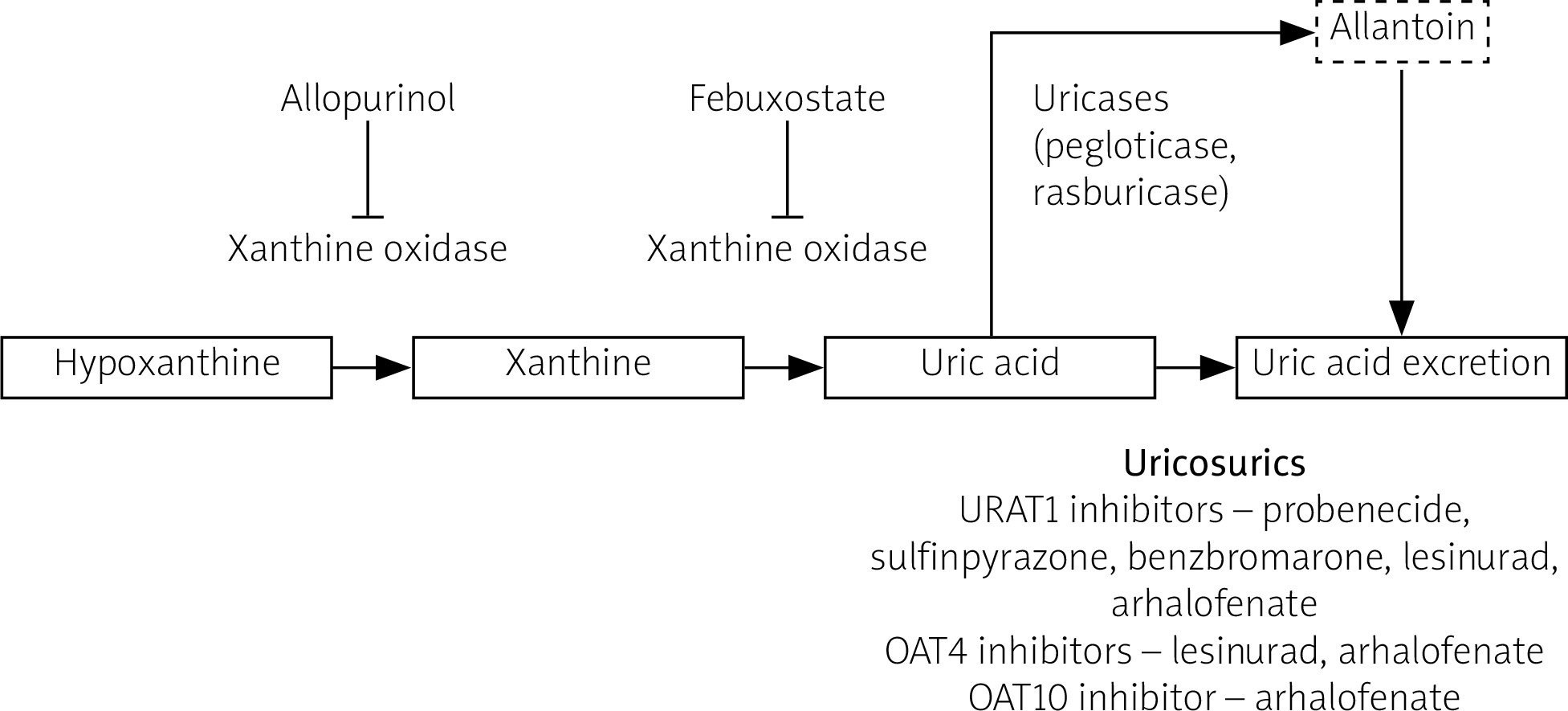
Similar to natural mammalian uricases, synthetic uricases transform UA to allantoin, a soluble form easily excreted with urine. In the case of inefficiency or intolerance of uricases, an interleukin 1 (IL-1) inhibitor (canakinumab or anakinra) may be administered, but only after excluding infection [23]. The mainstream UA lowering therapy drugs’ mechanisms of action are illustrated in Figure 2.
Fig. 2
Mechanism of action of urate-lowering drugs.
OAT4 – organic anion transporter 4, OAT10 – organic anion transporter 10, URAT1 – urate transporter 1.
Recommendations concerning colchicine and NSAIDs use in secondary gout prevention in patients with asymptomatic hyperuricemia differ in terms of suggested prophylaxis duration. It is agreed however that both colchicine and NSAIDs should be avoided in patients with severe renal dysfunction [20, 22, 23].
Also, colchicine should not be used in subjects receiving potent CYP3A4 inhibitors or P-glycoprotein inhibitors due to a high risk of drug interactions. Patients on long-term colchicine therapy, especially subjects with baseline renal dysfunction or on concomitant statin treatment, should be monitored for neuro- and myo-toxicity [23].
A common recommendation from rheumatological international societies is screening of patients with gout for comorbidities associated with hyperuricemia [20, 22, 23]. Conditions and factors mentioned in the “ACR 2012 Checklist” and EULAR guidelines are genetic and dietary factors, alcohol overuse, obesity, diabetes, hyperlipidemia, hypertension, metabolic syndrome, cardiovascular risk factors, chronic renal disease, nephrolithiasis, psoriasis, lead intoxication, lympho- and myeloproliferative diseases and use of some drugs [20, 23].
Medicaments potentially inducing hyperuricemia include diuretics, salicylates, cyclosporin, 2-ethylamino--1,3,4-thiadiazole, pyrazinamide, ethambutol, nicotinic acid and cytotoxic agents, as well as allopurinol or uricosurics in cases of too rapid UA lowering [35].
The EULAR guidelines do not address management of hyperuricemia in the case of co-existing comorbidities. Authors of “3E Initiative” indicate the need for renal function and cardiovascular risk factors’ evaluation in patients with gout but do not establish whether treatment of asymptomatic hyperuricemia in the presence of these comorbidities is necessary.
In the recent guidelines, ACR conditionally recommends against starting ULT in patients with asymptomatic hyperuricemia with or without comorbidities, based on the low incidence of gout in such patients noted in observational studies [21].
Hyperuricemia and cardiovascular disease
A large proportion of common hyperuricemia comorbidities are modifiable cardiovascular risk factors. It should be noted that recommended lifestyle modifications such as weight loss, regular exercise and dietary changes reduce both UA levels and the risk of gout, as well as insulin resistance [25]. Because of known associations between UA and cardiovascular risk, the question arises whether to apply ULT as a primary, secondary and/or tertiary prophylaxis of cardiovascular events.
Causes, clinical impact and treatment of asymptomatic hyperuricemia in patients with cardiovascular disease (CVD) remain controversial. High levels of UA in people suffering from CVD could be a result of reduced glomerular filtration rate, renal vasoconstriction, hyperinsulinemia, tissue ischemia, oxidative stress and/or diuretic treatment [36, 37]. On the other hand, many studies have proved that elevated UA is an independent risk factor of cardiovascular disease incidence and mortality [38].
One of the most thoroughly investigated issues is the correlation between hyperuricemia and arterial hypertension. The first observations were made on animal models. In rats, UA levels were proportional to blood pressure (BP) and UA lowering resulted in BP reduction [39]. In humans, UA leads to decreased nitric oxide production, increased creation of reactive oxygen species and renin–angiotensin system stimulation, which in turn causes renal vasoconstriction and UA-induced vasoreactive hypertension. These mechanisms apply to early-onset hypertension.
In the late stages of arterial hypertension, however, hyperuricemia is connected mainly with kidney microcirculation injury, and its direct association with increased BP values is less clear. The well-documented association between hyperuricemia and ischemic heart disease may be attributed to the aforementioned pathomechanism, as well as to increased platelet aggregation and in-stent restenosis [40].
The importance of UA in heart failure was mentioned in the recent European Society of Cardiology Guidelines [41]. Elevated UA level, mainly as a result of increased purine degradation linked to hypoxia, insulin resistance, as well as tissue catabolism (cachexia and sarcopenia), is an independent risk factor of poor prognosis, disease progression and mortality [42]. In heart failure patients with an implantable cardioverter-defibrillator (ICD), UA above 6.1 mg/dl may be a predictor of ventricular tachyarrhythmias requiring appropriate ICD therapies [43].
Allopurinol therapy reduces the risk of all-cause mortality [44], improves endothelial function [45] and lengthens walking distance in stable angina pectoris [46]. These effects do not always depend on lowering of the UA level. Prior to administration of xanthine oxidase blocking treatment, potential benefits and risk of side effects should be taken into consideration.
Cardiovascular disease screening in patients with elevated UA level and management of hyperuricemia in patients with cardiovascular risk are reviewed in detail in “Multidisciplinary experts consensus for the diagnosis and treatment of patient with hyperuricemia and high cardiovascular risk”.
According to experts’ opinion, the targeted UA level should be below 6 mg/dl in patients with hyperuricemia or below 5 mg/dl in patients with hyperuricemia and high cardiovascular risk (with at least two out of six risk factors: hypertension, dyslipidemia, diabetes, chronic kidney disease, history of myocardial infarction or stroke).
All patients with elevated UA level should receive adequate education on the role of diet and physical activity. Comorbidities should be assessed. Monotherapy with a xanthine oxidase inhibitor, usually allopurinol at the initial dose of 100 mg/day, should be considered as first-line treatment. In the case of ineffectiveness of low-dose allopurinol, increasing the dosage to 300–600 mg/day is recommended.
Once the UA level is normalized, ULT should be continued and UA level assessed every six months. In cases in which the therapeutic goal has not been achieved, a combined therapy may be considered. It is emphasized that febuxostat should not be recommended in patients with cardiovascular disease [47].
The role of uric acid in chronic kidney disease
The occurrence of hyperuricemia in chronic kidney disease patients is common. Serum UA level may be considered a marker of kidney disease and is increased mainly because of its impaired excretion. Results of studies investigating whether hyperuricemia itself may lead to the development of end stage renal disease are inconsistent.
It remains unclear whether an association between higher serum UA concentrations and CKD progression exists. A positive correlation has been recently described between hyperuricemia and development of diabetes mellitus, both types one and two [48, 49].
The authors of a large cohort study concluded that although elevated serum UA level correlates positively with the development of hypertension, it has no association with the incidence of chronic renal disease [50]. On the other hand, the authors of a review study stated that elevated UA is a predictor of development not only of CKD but also of AKI [51].
Recently, UA has been described as one of the most promising early markers of AKI. Compared to UA, increase of creatinine levels is delayed. Furthermore, creatinine levels are influenced by all-cause azotemia and falsely low in sepsis. Increased UA levels in AKI result from both increased UA production (in the course of ischemia) and decreased UA excretion (resulting from reduced glomerular filtration).
Uric acid is at the same time a biologically active indicator of intra-renal microcirculation injury, playing a role in the regulation of endothelial nitric oxide levels, inhibition of epithelial cell proliferation and migration, induction of endothelial cell apoptosis and inflammation through increasing monocyte chemoattractant protein 1 and C-reactive protein levels [52].
Although it has been hypothesized that hyperuricemia may be both a marker of CKD progression and a predictor of its development in the future, there are still no clear recommendations whether to treat asymptomatic hyperuricemia in the context of kidney disease prevention [51]. Results from a clinical trial showed a positive effect of allopurinol use on renal function, i.e. postponing the twofold serum creatinine level increase, eGFR decrease ≤ 50% and also the introduction of hemodialysis [53].
In a meta-analysis, Bose et al. [54] confirmed that ULT may reduce the prevalence of cardiovascular events, the most common cause of mortality in CKD, and ameliorate kidney disease outcomes. In some studies on the influence of febuxostat in CKD patients with gout, the therapy seemed to decrease the loss of renal function [55]. Authors of a more recent study showed however that while there may be a slight impact of allopurinol on lowering serum creatinine level in CKD, the effect of febuxostat was negligible [56].
Mortality in patients with chronic renal disease correlates with serum UA levels [57]. However, the evidence to make practical conclusions whether to treat high serum UA levels in CKD patients is still apparently insufficient, which is reflected by recommendations from Kidney Disease: Improving Global Outcomes (KDIGO), an international organization that issues guidelines for treatment of patients with kidney diseases [57].
Due to the modest data, the latest guidelines published in 2012 neither support nor oppose the use of ULT in patients with CKD in order to postpone the development of renal failure, regardless of whether the hyperuricemia is symptomatic or not.
On the basis of multiple studies, analyses and reviews, it seems that ULT in hyperuricemic CKD should be at least considered, especially in the context of other comorbidities. It appears that ULT may have a positive impact not only on CKD progression, but also on cardiovascular and survival outcomes.
Renal diseases directly related to uric acid and deposition of urate crystals
Renal diseases such as UA nephrolithiasis, as well as acute and chronic urate nephropathy, in which UA and crystal urate depositions are the cause rather than the effect of the pathology, require a separate therapeutic approach.
Acute urate nephropathy, defined as UA precipitation in renal tubules, develops most often due to tumor lysis syndrome (TLS) induced by radio- or chemotherapy received for myelo- and lymphoproliferative disorders. Plasma UA concentration in affected patients is usually above 15 mg/dl. The prevention of TLS in cancer patients is the gold standard.
Treatment with aggressive intravenous hydration and pharmacotherapy including allopurinol, febuxostat or rasburicase, or, in the most severe cases, hemodialysis, seems promising [58]. The treatment of this nephropathy in the course of other rare syndromes is more specific and goes beyond the scope of this paper.
On histopathology, chronic urate nephropathy is characterized by deposition of sodium urate crystals in the medullary interstitium. Since an acid urine pH and high UA concentration play the main role in pathophysiology of UA nephrolithiasis, treatment consists of urine alkalinization by potassium citrate or bicarbonate administration and a massive fluid intake. In the case of treatment failure or recurrence of nephrolithiasis, especially in patients with recurrent gout, xanthine oxidase inhibitors are recommended. It is worth mentioning that urine alkalinization and fluid therapy should not be discontinued after the introduction of a xanthine oxidase inhibitor.
Due to the fact that both nephrolithiasis and chronic urate nephropathy may have other underlying contributing factors, in each case a thorough investigation is necessary, especially to check for chronic lead intoxication and conditions leading to increased urine acidification.
Tumor lysis syndrome prevention and treatment
Very high UA levels (> 10 mg/dl in women and > 13 mg/dl in men) should be treated regardless of symptoms as they carry a serious risk of nephrotoxicity [3]. One of the reasons for significantly elevated UA levels is tumor lysis syndrome. Hyperuricemia (UA > 8 mg/dl or a 25% increase of UA from baseline) is one of the laboratory tumor lysis syndrome (TLS) classification criteria (alongside hyperphosphatemia, hyperkalemia and hypocalcemia) and an important prevention and therapy target.
Tumor lysis syndrome is an acute, life-threatening disorder occurring due to the breakdown of tumor cells accompanied by release of large amounts of nucleic acids – the source of UA, potassium and phosphates – into the bloodstream. It can lead to serious clinical manifestations including renal failure, cardiac arrhythmias and seizures [59].
Tumor lysis syndrome is predominantly induced by standard chemotherapy but can also be caused by radiotherapy, surgery or other targeted cancer treatment (such as hormone or antibody therapy) [60]. Occasionally, tumor lysis syndrome may occur spontaneously without treatment induction [59, 60].
The risk factors for TLS can be divided into two types: cancer-related (TLS most often occurs in hematological disorders) and unrelated to cancer. Risk factors of TLS unrelated to cancer include high baseline serum levels of UA, phosphates and potassium, co-existing kidney disease and male gender [18, 59].
According to current recommendations, pharmacologic prophylaxis of TLS including ULT and hydration is indicated in patients at intermediate and high risk of developing TLS and should begin at least 24 hours before the start of antitumor therapy [60].
In patients at low risk of TLS, ULT is not recommended, unless there are some additional risk factors such as those mentioned before [18]. Specific information on TLS risk classification and recommended TLS prophylaxis is included in Figure 3 [60].
Fig. 3
Tumor lysis syndrome prophylaxis [60].
ARF – additional risk factors, LDH – lactate dehydrogenase, UNL – upper norm limit, WBC – white blood cells.
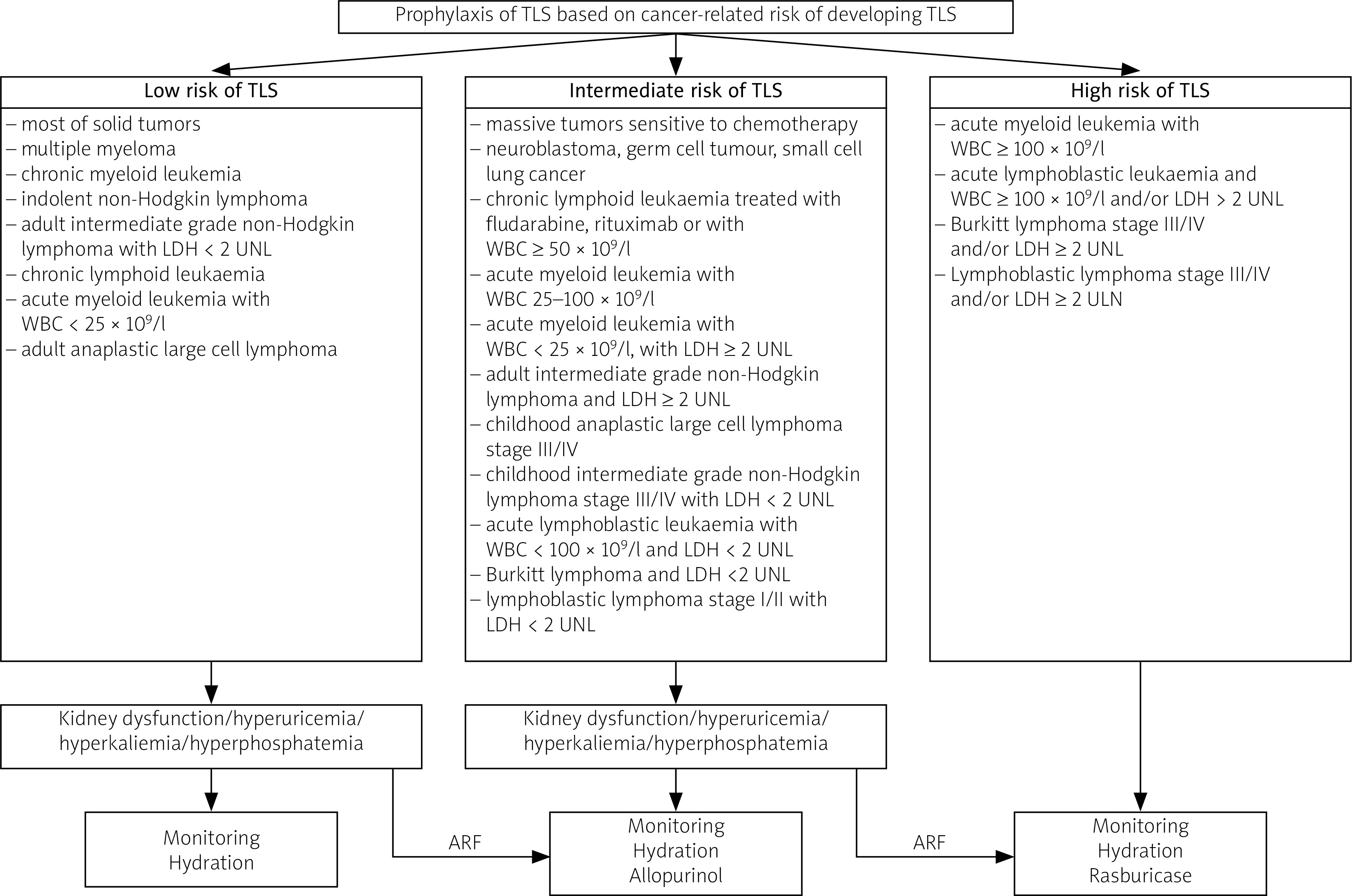
Agents suggested in TLS prophylaxis include allopurinol, febuxostat and rasburicase. To decide which agent should be prescribed for TLS prevention, some individual aspects should be taken into account. Allopurinol, although representing the first-line agent in TLS prophylaxis, does not lower levels of previously generated UA.
Its administration can lead to an increase in xanthine levels and subsequently to xanthine nephropathy. For this reason, allopurinol should be used only in patients at low or intermediate risk of TLS, whereas rasburicase should be the standard therapy for high-risk patients [60].
On the other hand, rasburicase should not be prescribed to patients with a history of glucose-6-phosphate dehydrogenase deficiency as it can provoke life-threatening hemolysis [59]. In the case of renal dysfunction or allopurinol intolerance, alternatively febuxostat may be used [59].
It also needs to be highlighted that prevention and management of TLS in some cancer patients can be especially difficult since ULT may be affected by several aspects of cancer disease such as renal failure, liver dysfunction and drug interactions. Allopurinol and azathioprine interaction can cause bone marrow suppression associated with an increased mortality risk.
Other drugs that may lead to serious drug interactions with allopurinol include cyclosporine, 6-mercaptopurine, cyclophosphamide, thiazide diuretics and amoxicillin [18, 60]. In patients receiving these drugs, selection, dosing and monitoring of proper ULT are crucial.
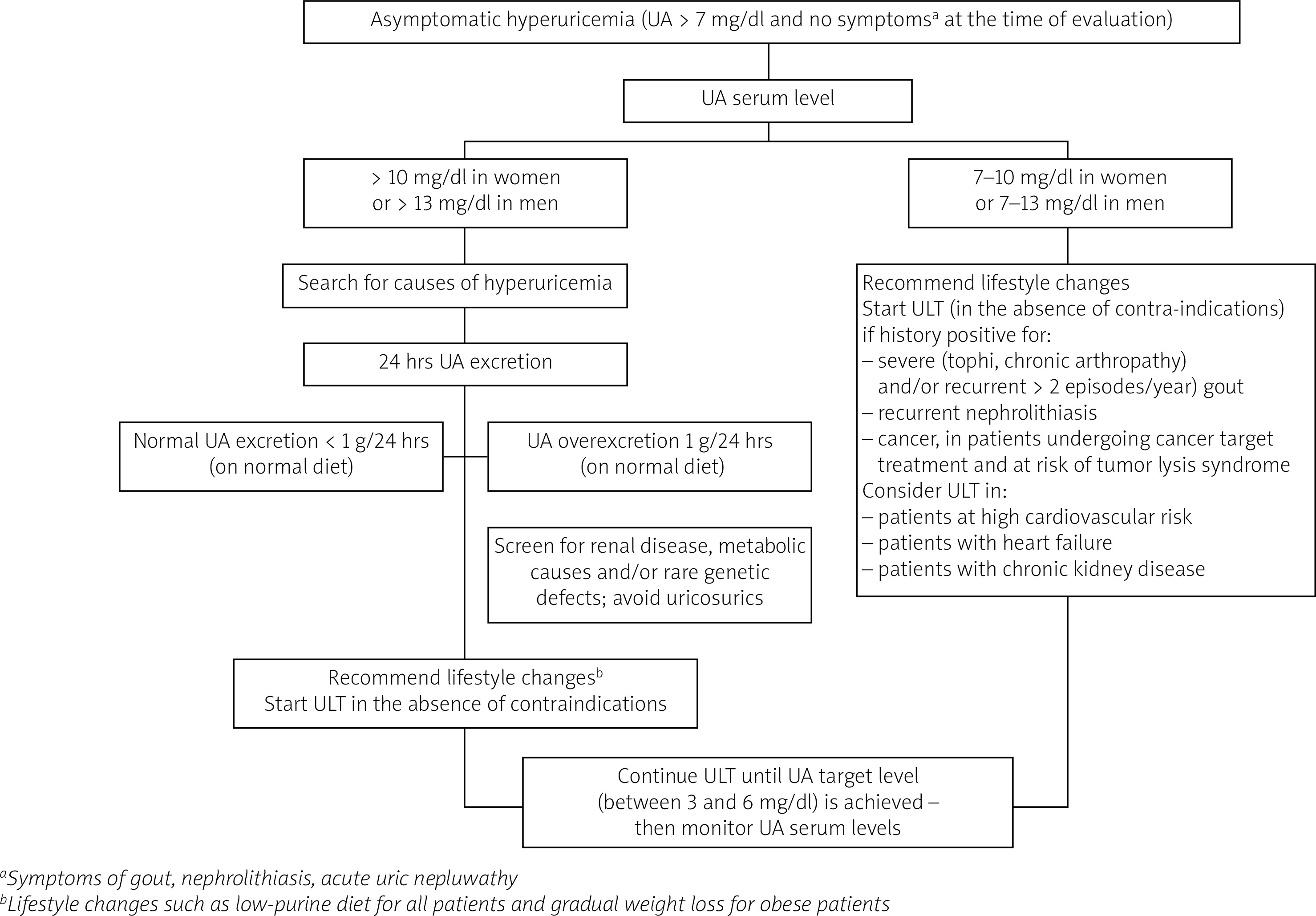
Hyperuricemia should be pharmacologically treated in patients with a history positive for severe or recurrent gout, nephrolithiasis or undergoing cancer treatment with an intermediate or high risk of tumor lysis syndrome, in order to prevent potential complications such as an episode of gout, renal colic or TLS.
Urate-lowering therapy should be administered and causes of hyperuricemia closely investigated in patients with especially high UA levels (> 10 mg/dl in women and > 13 mg/dl in men). An algorithm created based on data from studies presented in this review highlighting indications for ULT in hyperuricemia is presented in Figure 4.
Conclusions
Routine testing for serum UA level should be undertaken especially in patients with a family history of gout, nephrolithiasis or early cardiovascular disease, as well as in patients treated for cancer. Increased UA concentration may help identify patients at increased risk of gout, diabetes, dyslipidemia, hypertension, metabolic syndrome, cardiovascular disease and chronic renal disease.
Regular screening for these conditions in hyperuricemic patients may help in establishing appropriate preventive interventions, early diagnosis and treatment. Uric acid level may also be a relevant prognostic tool in the clinical monitoring of chronic kidney disease, heart failure and cardiovascular disease progression, as well as a predictor of disease mortality in these disorders.
Urate-lowering therapy probably should be administered to patients with chronic kidney disease, diabetes, cardiovascular disease, heart failure or a history of a cardiovascular event in order to lower mortality associated with high cardiovascular risk. However, there is no current international consensus on how to manage hyperuricemia in these patients. Studies on long-term effects of ULT in patients burdened with high cardiovascular risk are warranted in order to draw final conclusions.
The UA threshold for starting ULT in hyperuricemic patients may differ depending on the patient’s history and present disorders. A treat-to-target approach aiming to reach levels below the UA crystallization threshold with a safe margin, usually below 6 mg/dl, seems to be reasonable, regardless of the reason for ULT introduction.
Attention should be paid to ULT monitoring in order to avoid decreasing UA concentration below the lower value of the targeted UA level range, that is 3 mg/dl. Uric acid seems to play some important physiological, e.g. neuroprotective, functions, whose mechanisms remain unclear.