
Introduction
Felty’s syndrome (FS) is a rare systemic connective tissue disease. It is characterised by a triad of symptoms: seropositive rheumatoid arthritis (RA), splenomegaly and neutropenia. Splenomegaly does not occur in all patients. To establish the diagnosis of FS, it is necessary to meet the classification criteria for RA and have the presence of neutropenia, indicated by a neutrophil count in the peripheral blood below 1.5 × 103/μl [1].
The underlying cause of neutropenia in the progression of FS is not entirely clear. It may result from various factors, including splenomegaly, which leads to sequestration and destruction of neutrophils, bone marrow suppression caused by interferon γ and peripheral destruction caused by autoantibodies targeting neutrophils and granulocyte growth factor (G-CSF) [2].
The course of FS can manifest as an asymptomatic condition. There are no active cases of peripheral arthritis, neutrophil splenomegaly or related concomitant infections. However, FS typically affects patients with long-standing RA and active arthritis. Occasionally, symptoms such as splenomegaly and neutropenia may already be present when RA is diagnosed.
The influence of two alleles of the HLA-DRB1*04 gene, which predispose individuals to the development of the disease, is indicated in the aetiopathogenesis of FS. The presence of the HLA-DRB1*04 gene alleles in a homozygous system increases the risk more significantly compared to the heterozygous system [1, 2].
Clinically, FS is most commonly manifested by active peripheral arthritis and both generalised and localised infections. Infections predominantly affect the respiratory system and skin. Simultaneous use of immunosuppression can potentially lead to life-threatening infectious complications [1].
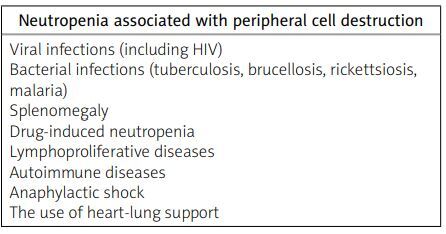
When considering the differential diagnosis, it is important to include diseases that may cause neutropenia. These diseases can be categorized into congenital and acquired conditions (Table I). Neutropenia can arise from various causes, including impaired production of neutrophils (Table II), increased destruction of granulocytes in the peripheral blood (Table II), and improper distribution of granulocytes in the peripheral blood.
Table I
Differential diagnosis of impaired granulocyte production based on [3]
Table II
Causes of neutropenia related to peripheral cell destruction based on [3]
In addition to FS, other conditions that may present with isolated granulocytopenia and splenomegaly include storage diseases, chronic infections (such as tuberculosis, sarcoidosis, and bacterial endocarditis), systemic lupus erythematosus, and, rarely, Sjögren’s syndrome. The risk of infection increases as the duration of granulocytopenia lengthens. Infections resulting from neutropenia usually do not occur when the granulocyte count is maintained between 1 × 103/μl and 1.5 × 103/μl [3].
The treatment approach for FS typically involves the use of conventional synthetic disease-modifying drugs (DMARDs) as well as biological DMARDs (bDMARDs) [1, 4, 5]. Biological drugs employed in FS treatment include TNF-a inhibitors (such as etanercept, infliximab and adalimumab) and anti-CD20 antibodies (such as rituximab).
A comparative analysis of 8 patients treated with rituximab and 6 patients treated with a TNF-α inhibitor revealed that rituximab exhibited improved efficacy. Patients treated with rituximab demonstrated positive clinical responses, characterized by increased neutrophil counts, reduced inflammation markers and decreased Disease Activity Score 28 (DAS28). Among this group, only 1 out of 8 patients experienced a recurrence of neutropenia during long-term follow-up.
Conversely, patients treated with anti-TNF-α agents did not achieve the desired therapeutic goals in most cases, with only a few patients demonstrating improvement in DAS28 without resolving neutropenia [6]. Consequently, when classic synthetic DMARDs prove ineffective or lose their efficacy, the preferred course of action is to initiate rituximab as the first-line treatment option.
Splenectomy can also be used to treat neutropenia; however, its indications in FS are limited. It may be considered in cases of a poor response to conventional synthetic and biological DMARDs, persistent neutropenia and recurrent infections [1].
Material and methods
A systematic search of the literature was conducted on the electronic database PubMed using the keywords “Felty’s syndrome”, “treatment”, “neutropenia”, and “clinical symptoms” within the timeframe of January 1990 to April 2023. The aim of this article was to analyse the possibility of biological treatment in FS.
Results
During the specified time period and considering the subject of publication, a total of 385 articles or book chapters related to FS were found (Fig. 1).
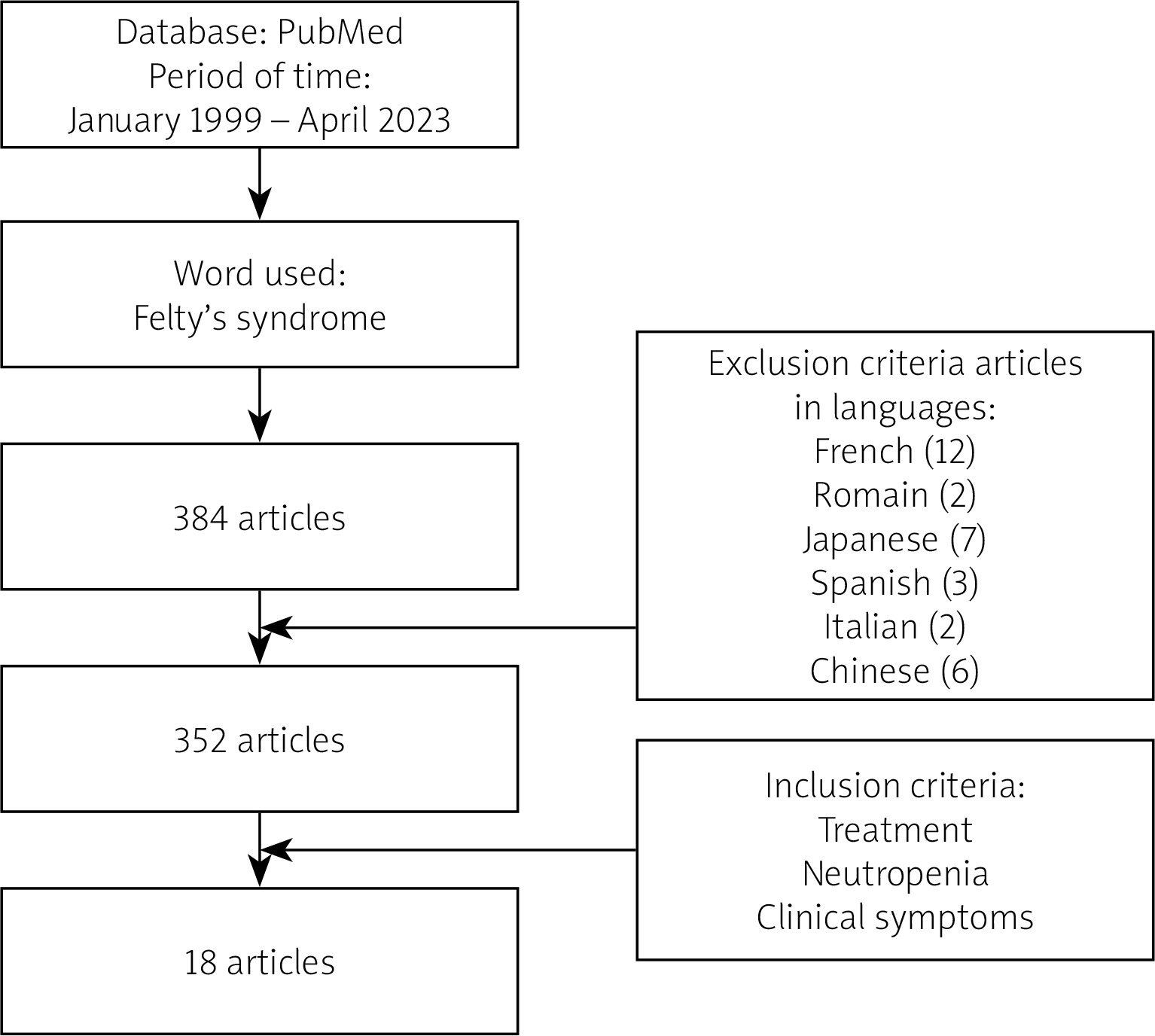
Out of the initially identified 385 articles 334 were excluded from the analysis, resulting in a final selection of 18 articles that fulfilled the predetermined criteria. A majority of the articles meeting the criteria were either case reports or repetitions of previously published papers.
Based on the analysis of the literature, it was found that there is currently no universally accepted treatment regimen for FS. The therapies employed are primarily based on the treatment of RA and the experiences of various rheumatology centres (Table III).
Table III
Biological treatment in Felty’s syndrome based on avialable literature
| First author and year of publication | Biologic treatment | Treatment effectiveness |
|---|---|---|
| Ghavami et al. 2005 [7] | Etanercept | Partial |
| Kimura, Yoshida 2020 [8] | Abatacept | Successful |
| Li et al. 2020 [9] | Rituximab | Successful |
| Gupta et al. [10] | Adalimumab | Partial |
| Li et al. 2020 [11] | Tocilizumab | Successful |
| Tomi et al. 2012 [12] | Rituximab | Successful |
| Chandra et al. 2008 [13] | Rituximab | Successful |
| Wang 2018 [14] | Rituximab | Successful |
| Shipley et al. 2008 [15] | Rituximab | Successful |
| Lekharaju, Chattopadhyay 2008 [16] | Rituximab | Successful |
| Becker et al. 2014 [17] | Rituximab | Successful |
| Weinreb et al. 2006 [18] | Rituximab | Successful |
| Heylen et al. 2012 [19] | Rituximab | Successful |
| Pukšić et al. 2017 [20] | Rituximab | Successful |
| Sordet et al. 2005 [21] | Rituximab | Ineffective |
| Ayzenberg, Shenberge 2014 [22] | Rituximab | Successful |
| Salama et al. 2008 [23] | Rituximab | Successful |
| Sarp, Ataman 2014 [24] | Rituximab | Successful |
The case described below effectively highlights the difficulties encountered in the treatment of FS.
Case description
A 45-year-old male patient with no significant medical history was urgently admitted to the Department of Internal Medicine in February 2018 due to exhibiting polyarthritis and splenomegaly. On admission, leukopenia (1.31 × 103/μl), mild normocytic anaemia, neutropenia (0.51 × 103/μl) along with lymphopenia (0.54 × 103/μl), increased CRP concentration (16.57 mg/l), presence of rheumatoid factor at low titre (23 IU/ml) and ANA at a titre 1 : 320 were observed. The laboratory test results upon admission, as well as throughout the entire follow-up, can be found in Table IV.
The abdominal ultrasound revealed splenomegaly measuring up to 192 mm (normal value up to 160 mm). In order to assess the neutrophil marrow reserve, a hydro-cortisone test was conducted [3]. This involved intravenous administration of 100 mg of hydrocortisone, followed by the evaluation of the neutrophil count in the peripheral blood after 4 and 6 hours. The results of the hydrocortisone test did not show the required increase in neutrophils of at least 2 × 103/μl.
Consequently, a bone marrow aspiration biopsy was performed to investigate the underlying cause of the observed leukopenia and neutropenia. Based on the biopsy results, a proliferative process was ruled out as the cause. Due to the suspicion of FS, the patient was subsequently transferred to the Rheumatology Department for further diagnostics.
During the hospitalisation at the Rheumatology Department, clinical examination revealed evident signs of active, symmetrical peripheral arthritis. The Disease Activity Score 28 (DAS28) was measured at 6.5, indicating high disease activity. These findings were further confirmed by joint ultrasound imaging.
Additionally, laboratory tests showed the presence of anti-citrulline antibodies (ACPA) at a titre exceeding 200 U/ml (normal value: 0–5 U/ml). However, no detectable levels of extractable nuclear antigen antibodies (ENA) or anti-dsDNA antibodies were found in the laboratory tests.
Based on the patient’s fulfilment of the 2010 criteria established by the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR; now European Alliance of Associations for Rheumatology) for RA [25], along with the presence of splenomegaly and neutropenia below 1.5 × 103/μl, the patient was ultimately diagnosed with FS.
Due to severe leukopenia with neutropenia and lymphopenia, the patient was disqualified from initiation of methotrexate (MTX) therapy as recommended by the consulting haematologist. Instead, the treatment approach involved the use of glucocorticosteroids (GS) and hydroxychloroquine (HCQ) at a daily dose of 200 mg.
Despite the therapy (administered from February 2018 to May 2019), it was not possible to reduce the GS (prednisone) dosage below 20 mg/day. Persistent symptoms of peripheral arthritis (DAS28 greater than 5.1), increased markers of inflammation and ongoing neutropenia requiring periodic use of granulocyte growth factors and antibiotic prophylaxis. Due to persistent leukopenia with neutropenia, splenic artery embolization was performed in March 2019. However, despite this intervention, the anticipated increase in the number of neutrophils in the peripheral blood did not occur as expected.
During a follow-up ophthalmologic examination, pigment rearrangements in the macula and multiple retinal pigment epithelial defects were revealed. As a result, HCQ had to be discontinued. In July 2019, cyclosporine A (CsA) was initiated at a daily dose of 50 mg, but it had to be discontinued after ten days due to an increase in the patient’s blood pressure. With CsA and HCQ no longer viable options, it was decided to initiate MTX therapy.
After consulting with the haematologist, treatment began in August 2019 with a weekly dose of 10 mg. Unfortunately, this dosage failed to achieve the intended therapeutic goal. High disease activity (DAS28 > 5.1) persisted, preventing a reduction of the doses of GC used so far to less than < 15–20 mg of prednisone per day. As a result, the patient was deemed eligible for a biological treatment programme involving a TNF-α inhibitor (etanercept).
After 3 months from the start of receiving etanercept therapy, significant clinical improvement was observed with a decrease in disease activity (DAS28 < 3.2) and laboratory improvement. As a result, GC were discontinued. After eight months of etanercept treatment, the patient experienced a recurrence of leukopenia with neutropenia and developed thrombocytopenia with platelet count in the range 90–130 × 103/μl (normal range: 150–450 × 103/μl).
Several attempts were made to discontinue etanercept, which led to subsequent increases in platelet count. However, each time the patient resumed etanercept therapy, the platelet count dropped below the normal range. Finally, it was decided to discontinue the etanercept treatment, and the patient was qualified for anti-CD20 antibody therapy (rituximab) in December 2020. The patient responded well to this modification of treatment, achieving remission of the underlying disease.
Clinical signs of active arthritis were not observed, and inflammation parameters remained low. Follow-up peripheral blood tests showed an increase in leukocyte count, stabilization of neutrophil count at the range of 1–2 × 103/μl, and normalization of platelet count at the range of 170–200 × 103/μl. The patient no longer required GS therapy.
Discussion
Some studies have suggested that the initial treatment for FS should involve low-dose MTX [5, 26, 27]. Other DMARDs, including leflunomide, HCQ, CsA, sulfasalazine, azathioprine, and cyclophosphamide, have also been used to treat FS [4, 28–30].
Considering the characteristics of the MTX medical product, leukopenia is listed as one of the contraindications for its use. Neutropenia, on the other hand, may occur as a result of drug-induced myelosuppression. Persistent leukopenia < 3 × 103/μl is an indication for reducing MTX dosage.
Therefore, when using MTX in the treatment of FS, it is crucial to exercise extreme caution and closely monitor haematological parameters. In the case described, an attempt was made to administer MTX; however, due to the lack of clinical improvement and increasing neutropenia, it was decided to include biological drugs in the treatment regimen.
At that time, bDMARDs were considered a safer approach. The patient responded well to treatment with a TNF-α inhibitor (etanercept), resulting in a decrease in disease activity and normalization of peripheral blood neutrophil counts. However, during the therapy, there was a gradual decline in platelet count, stabilizing within 90–130 × 103/μl (normal range: 150–450 × 103/μl). This level of platelet count meets the definition of thrombocytopenia.
Thrombocytopenia is listed as a side effect of the two most commonly used etanercept preparations in our centre. According to the product information, this complication is considered “uncommon”, occurring in approximately 1 in 1,000 patients [31, 32].
In 2009, a study was published on thrombocytopenia occurring in patients with cutaneous or joint psoriasis who were being treated with TNF-α inhibitors, including infliximab, adalimumab, and etanercept. The study involved 93 patients, and among them, 4 patients exhibited laboratory findings of thrombocytopenia, which was defined as a decrease in platelet count below 50 × 103/μl. The study highlighted the immunological basis of this complication, as antiplatelet antibodies and antinuclear antibodies were detected in the serum of these patients.
Moreover, the patients showed a positive response to treatment with GCs. In 1 patient, thrombocytopenia was diagnosed incidentally during treatment with etanercept. In the remaining 3 patients, thrombocytopenia was diagnosed after the onset of clinical symptoms of thrombocytopenia. A spontaneous increase in platelets was also observed upon discontinuing TNF-α inhibitor therapy [33].
After switching from a TNF-α inhibitor (etanercept) to an anti-CD20 antibody (rituximab), our patient responded well to the treatment. There were no signs of active arthritis, inflammation parameters were low, and thrombocytopenia was not observed. Other authors have also reported positive outcomes in the treatment of FS using rituximab. They have described favourable experiences with rituximab therapy, including a decrease in disease activity measured by the DAS28 index and normalization of neutrophil levels in peripheral blood.
A study that followed 9 FS patients for a period of 15 years reported similar findings. These patients received rituximab either as a standalone treatment or in combination with other conventional DMARDs. In all cases, rituximab therapy led to a decrease in disease activity and the restoration of normal neutrophil levels in peripheral blood. These effects were observed regardless of baseline disease activity and duration [9, 12–20, 22–24].
In 2020, successful treatment of FS using tocilizumab [11] or abatacept [8] was also reported. These findings suggest that they can be considered as potential therapeutic options for FS.
In the context of FS, the decision to use granulocyte growth factor (G-CSF) is crucial due to the risk of infectious complications associated with neutropenia. These complications are the leading cause of mortality in FS patients. However, there are no clear criteria for qualifying patients for G-CSF administration as a preventive measure of febrile neutropenia. Haematological recommendations determine the use of G-CSF based on the patient’s risk group for febrile neutropenia: low, intermediate, or high.
Assignment to risk groups for febrile neutropenia is based on specific chemotherapy regimens, not the absolute neutrophil count. Specific chemotherapy regimens determine the patient’s risk group for febrile neutropenia.
For non-oncology patients, general risk factors and the MASCC (Multinational Association for Supportive Care in Cancer) prognostic index are used to assess the risk [34]. Granulocyte colony-stimulating factor effectively treats neutropenia but may also lead to worsening joint symptoms, which may be related to the release of IL-6 [35].
Conclusions
Felty’s syndrome being a systemic connective tissue disease requires careful selection of appropriate pharmacotherapy by physicians. Treatment minimises the risk of irreversible joint damage caused by active inflammatory arthritis.
Awareness of the potential occurrence of rare but possible side effects of medications is crucial. It is also important to understand the potential complications associated with the use of DMARDs.