
Introduction
Cushing syndrome (CS) is a consequence of prolonged exposure to excessive amounts of circulating glucocorticosteroids (GCs). Chronic administration of GCs is the most common cause of CS while pituitary neuroendocrine tumour (PitNET) secreting adrenocorticotropic hormone (ACTH), known as Cushing disease (CD), remains the most frequent cause of endogenous cortisol hypersecretion, followed by ACTH-independent cases (predominantly adrenal adenoma) and ectopic corticotropin-releasing hormone (CRH)/ACTH-producing neoplasms (e.g., bronchial neuroendocrine neoplasms) [1]. The annual incidence of CS ranges between 1.8 and 3.2 cases per million [1–3].
Despite scientific progress since 1932, when Harvey Cushing described the first case of the clinical syndrome that now bears his name, the diagnosis of endogenous CS is often delayed (mean time to diagnosis 34 months) and poses a significant diagnostic challenge [4]. Cushing syndrome is characterized by increased mortality and various comorbidities with a pronounced impact on quality of life, e.g., metabolic and neuropsychiatric disorders, impairment of reproductive and sexual function, dermatological manifestations, but also musculoskeletal complications [5]. The latter including osteoporosis, skeletal fractures, and myopathy [5, 6].
Cushing syndrome may damage the musculoskeletal system both structurally and functionally and the level of impairment depends mainly on the time of exposure and excessive concentration of GCs [7]. Although endogenous CS remains a rare entity, GCs are among the most frequently prescribed drugs: the weighted prevalence of oral GC use was 1.2% between 1999 and 2008 in the United States [8]. Interestingly, only 37.9% of oral GCs users reported concomitant use of any anti-osteoporotic agent (bisphosphonates, calcitonin, calcium, hormone replacement therapies, teriparatide, and vitamin D) [8]. Thus, early identification of patients at risk of musculoskeletal complications of excess of GCs and adequate treatment remains essential.
In this review, we discuss the pathophysiology, clinical presentation, prevention, and management of musculoskeletal complications of prolonged exposure to GCs.
Skeletal complications of glucocorticosteroids excess
Epidemiology
Any chronic excess of GCs, regardless of cause, leads to bone loss and increased risk of fractures. Deterioration of bone status has been described in 64–100% of patients with endogenous CS, osteopenia occurs in 40–78%, osteoporosis in 22–57%, and skeletal fractures in 11–76% of patients [5]. More than 10% of patients who receive long-term treatment of GC are diagnosed with a clinical fracture, and 30–40% have radiographic evidence of vertebral fractures [9].
The risk of fractures induced by oral GCs is related mainly to the daily dose, not to the cumulative dose of GCs [10]. In endogenous CS, bone status largely correlates with severity of hypercortisolemia; therefore patients with ectopic ACTH secretion have lower bone mineral density (BMD) and a higher risk of fractures compared to patients with pituitary or adrenal CS [11]. Patients with adrenal hypercortisolemia experience more severe bone loss than patients with CD because of the protective role of adrenal androgens (including dehydroepiandrosterone sulfate – DHEA-S), which are higher in CD because of ACTH stimulation [12, 13]. Men with endogenous CS are more likely to suffer from osteoporosis and vertebral fractures than women, which suggests that testosterone deficiency additionally negatively affects the bone condition in CS [14].
The excess of GCs impairs mainly the trabecular bone, leading to an increased risk of osteoporotic fractures mainly of the spine, which are often the first symptom of CS. The results of the Danish Cohort Study showed that the risk of fractures of patients with endogenous CS increased particularly within the 3 years prior to diagnosis and treatment and became lower with longer follow-up duration [15]. These data indicate that endogenous CS is an insidiously evolving disease. Therefore, prompt diagnosis and treatment of CS are crucial to limit bone damage. In exogenous GCs excess the highest rate of bone loss occurs predominantly within the first 3–6 months of GCs treatment, even before significant loss of BMD, and a slower decline continues with persistent use [16].
Pathophysiology – direct effects of glucocorticosteroids on bone status
Knowledge on the effect of GCs on the bone status through direct and indirect mechanisms and the pathogenesis of GC-induced osteoporosis (GIO) comes mainly from studies of patients treated with exogenous GCs and experimental animal model testing.
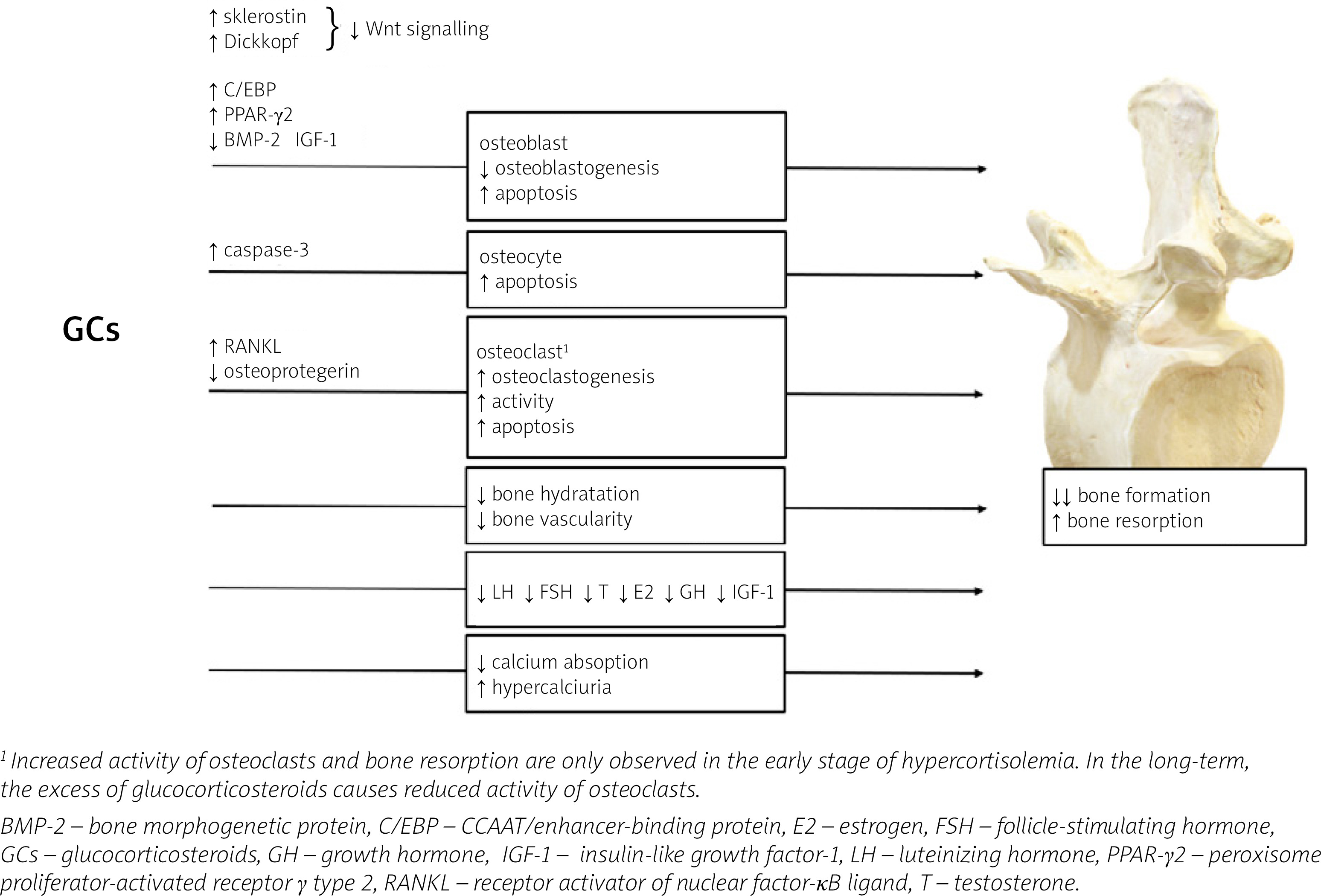
At the tissue level, GCs mainly disturb bone formation processes, in contrast to postmenopausal osteo-porosis or hyperparathyroidism, in which bone resorption dominates. The decrease in bone formation is the result of inhibition of osteoblast differentiation, function and lifespan which is attributed to inactivation of the Wnt/β-catenin signaling pathway (a critical pathway for osteoblastogenesis), the induction of nuclear factors of the CCAAT/enhancer-binding protein (C/EBP) family and peroxisome proliferator-activated receptor γ (PPAR-γ) type 2, as well as repression of bone morphogenetic protein (BMP-2) [17]. Glucocorticosteroids may also enhance bone marrow stromal cell development toward the adipocyte lineage rather than toward osteoblasts [18].
In addition, GCs also promote apoptosis in osteoblasts and osteocytes due to activation of caspase-3 [19] and may also affect the metabolism and function of osteocytes, modifying the elastic modulus surrounding osteocyte lacunae and causing reduced mineral-to-matrix ratios in the same areas with an increase in lacunar size [20]. These effects of GCs on osteoblasts and osteocytes might account for a disproportionate loss of bone strength in relation to bone mass. In fact, in both endogenous and exogenous hypercortisolism, fractures may develop even in the presence of normal or low-normal BMD, measured by dual-energy X-ray absorptiometry (DXA) [21].
Apart from the effect on osteoblasts and osteocytes, GCs can also intensify the process of bone resorption by disturbing the balance between receptor activator of nuclear factor-κB ligand (RANKL) and osteoprotegerin (OPG). RANKL produced by osteoblasts and osteocytes, is essential for osteoclast maturation, activation and survival [22, 23]. The effects of RANKL are counteracted by osteoprotegerin, a neutralizing decoy receptor which thus inhibits maturation and differentiation of the osteoclasts. The balance between RANKL and OPG during GCs treatment is disturbed and favors of RANKL leading to an initial increase in bone resorption. However, owing to the potent suppressive effects of GCs on osteoblasts, the increase in RANKL is only transient [24].
Glucocorticosteroid-induced bone loss occurs in two phases: a rapid, early phase in which bone mass is lost due to excessive bone resorption and a slower, later phase in which bone is lost due to inadequate bone formation. These two phases of action of GCs were confirmed by Yao et al. [25]: in this study, gene expression and changes in bone microarchitecture were assessed in GC-treated mice on days 0, 7, 28 and 56: GC excess induced early up-regulation of genes involved in osteoclast activation, function, and adipogenesis, which peaked on day 7; from day 28 to 56, genes associated with osteoblast maturation and activation significantly decreased from the baseline values, and Wnt/β-catenin antagonists including Dickkopf-1 and sclerostin were increased. These changes in gene expression were consistent with changes in the biochemical markers of bone turnover and bone mass, with an initial increase in bone resorption markers such as serum cross-linked C-telopeptide of type I collagen (CTX-I) and a reduction in trabecular bone volume of the spine, followed by a decrease in the bone formation marker osteocalcin [25].
Furthermore, studies in GC-treated mice have shown a decrease in bone hydration accompanied by a reduction in bone vascularity and blood flow compared to control mice, which may be an additional mechanism whereby GCs reduce bone strength [26, 27]. Moreover, GCs mediate changes in synthesis and binding of receptors or growth factor binding proteins present in the bone microenvironment [17]. Glucocorticosteroids inhibit the expression of IGF-1 in osteoblasts, which stimulates bone formation and type I collagen synthesis and reduces bone collagen degradation and osteoblast apoptosis [28].
One of the features of GIO that is still not sufficiently explored is the high interindividual variability of its clinical course and severity. This variable effect of GCs on bone status may be the result of the glucocorticosteroid receptor (GR) polymorphisms associated with increased (BclI and N363S) or decreased (ER22/23EK and A3669G) susceptibility to GCs or possibly local 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) activity [29, 30]. Osteoblastic 11β-HSD1 can effectively convert prednisone to active prednisolone with kinetics similar to cortisone to cortisol conversion [31]. This suggests that osteoblastic 11β-HSD1 may influence the sensitivity of bone to therapeutic GCs.
Glucocorticosteroids increase the activity of 11β-HSD1, but also pro-inflammatory cytokines, which significantly induces the sensitivity of bones to GCs in the course of an inflammatory diseases [32].
Indirect effects of glucocorticosteroids on bone status
Indirect effects of GCs on bone status include mainly disturbances in the secretion and action of hormones. Both exogenous and endogenous GC excess inhibit the hypothalamic–pituitary–gonadal axis. Decreased testosterone and estrogen production may be an additional factor in the pathogenesis of GIO [33].
Glucocorticosteroids also inhibit the IGF-1 axis (GH–IGF-1). Growth hormone directly and through IGF-1 stimulates the process of bone formation [17]. Functional GH deficiency was observed even when the GCs excess was mild, as in patients treated with inhaled GC and in those with “subclinical” endogenous hypercortisolism [17, 34, 35].
Another indirect mechanism for the adverse skeletal effects of GCs is a decrease in intestinal calcium absorption as a result of inhibition of vitamin D actions, decrease of the expression of specific calcium channels in the duodenum and increase in renal calcium excretion. Physiologically in this situation one would expect to find evidence of a secondary increase in parathormone (PTH). However, no significant change in PTH levels was found in most CS patients.
Furthermore, direct histomorphometric analysis of bone biopsies demonstrated reduced bone turnover in CS, in contrast to the increased bone turnover that characterizes hyperparathyroidism [25]. This indicates a relatively minor role of PTH in the bone loss associated with GCs use. Nevertheless, GCs have a significant effect on the dynamics of PTH secretion, with a decrease in tonic PTH release and an increase in pulsatile releases of this hormone [36]. In addition, GCs have been reported to increase the expression and availability of PTH receptors on osteoblasts, which may be associated with increased sensitivity of skeletal cells to PTH [37].
The mechanisms of GCs excess action on bone are summarized in Figure 1.
Clinical presentation
Given that GIO is an example of a bone disorder in which fractures often develop in patients with normal or low normal BMD, standard DXA alone is not a sufficient tool to assess bone status in CS, in which, mainly bone quality is impaired. Bone microarchitecture, probably the most important determinant of bone quality, may explain bone fragility in CS.
It was originally studied by histomorphometry, but it can currently be evaluated by computed tomography systems [high resolution peripheral quantitative computed tomography (HRpQCT) and central quantitative computed tomography (QCT)] or micromagnetic resonance imaging (microMRI), which are not readily available in a clinical setting [38]. A non-invasive technique that correlates with HRQCT and QCT measurements and has gained acceptance in assessing bone micro-architecture is the trabecular bone score (TBS) [39]. The lumbar spine TBS is a grey-level texture measurement based on the use of experimental variograms that can be extracted from the two-dimensional lumbar spine DXA image. Low TBS values are associated with thin trabeculae, low trabecular number, and low connectivity density, which reflects poor micro-architectural bone quality [40].
Recent studies showing patients receiving GCs have lower TBS Z-scores than GCs-naïve patients without differences in their BMD Z-scores [41, 42].
In addition to the assessment of bone quality, morphometric vertebral assessment is recommended in patients with CS. Vertebral fractures are often overlooked, being frequently asymptomatic [43]. Height loss greater than 3–5 cm is an indicator of possible pre-existing vertebral fractures [44].
Treatment
Most studies have shown that GC bone loss and GIO are potentially reversible after successful surgical treatment, although the time to bone recovery is relatively long and variable and restoration is not always complete [45–49]. An improvement in BMD was noted 3 to 6 months after hypercortisolism remission, generally slower at the femoral neck than at the lumbar spine [46, 49]. A long-term prospective study showed normalization of BMD at the lumbar spine and femoral neck in patients with endogenous CS after successful surgical treatment after a mean follow-up of 71 months [45]. While in endogenous CS, resolution of the underlying disease can resolve the problem of bone damage, in exogenous GIO, patients are often unable to taper off therapeutic doses of GC.
Oral administration of each GC in a dose equivalent to 2.5 mg of prednisone or higher daily over 3 months or longer increases the risk of fracture [50]. Previous studies suggested a less negative impact on bone status of deflazacort than other GCs [51, 52]. However, a recent retrospective study on GC-treated boys with Duchenne muscular dystrophy (DMD) reported higher fracture risk of the group treated with daily deflazacort compared with the group treated with daily prednisolone [53].
The severity of GIO is less pronounced in patients treated with high dose 6-methylprednisolone intravenous pulse therapy than daily oral GC therapy [54]. It is noteworthy that even high dose of inhaled GC (≥ 1,000 μg of fluticasone daily or equivalent) for more than 4 years increases the risk of fracture [55].
All orally administered GCs (intermediate- and long-acting) should be administered in a single morning dose to adjust to the physiological cortisol circadian rhythm. However, the use of hydrocortisone (short-acting GC) in doses higher than 30 mg in replacement therapy in adrenal insufficiency was proven to induce bone loss [56].
All patients exposed to excess GCs should be treated with adequate vitamin D and calcium supplementation. Lifestyle modification (balanced diet, smoking cessation, regular weight-bearing or resistance training exercise) is also necessary. The guidelines recommend 1,200–1,500 mg of calcium daily, including all sources (supplements, dietary intake). The clinical goal of vitamin D supplementation is to achieve a minimum concentration of 30 ng/ml [57].
In addition to correcting concomitant risk factors for fractures, treatment with anti-osteoporosis drugs may be necessary in persistent endogenous CS. However, evidence-based knowledge on the use of anti-osteoporosis treatments in endogenous CS is scarce and no specific guidelines are available in this area.
In contrast to this group of patients, there are specific guidelines for exogenous GIO that can be only partially translated to patients with endogenous CS [9, 58]. The guidelines recommend modulation of treatment with exogenous GIO depending on individual fracture risk, which should be assessed in all patients receiving prednisone (or equivalent) at a daily dose > 2.5 mg for > 3 months. The most important elements of this evaluation are age, previous history of fractures, daily dose and duration of GCs used and individual fracture risk profile (calculated by the Fracture Risk Assessment Tool – FRAX). Oral bisphosphonates are recommended in patients at moderate to high risk of fracture and in any patient with prevalent fracture.
According to Polish recommendations, this procedure should also be initiated obligatorily in patients over 65 years of age, receiving ≥ 7.5 mg/day prednisone, even if they present no other fracture risk factors [59]. Oral bisphosphonates are preferred as first-line drugs due to safety, cost, and lack of evidence for superior anti-fracture effects of other anti-osteoporosis medications, in accordance with the relevant guidelines.
Other therapies are recommended if oral bisphosphonates are not appropriate, in order of preference: bisphosphonates intravenously, teriparatide, denosumab (anti-RANKL monoclonal antibody) and raloxifene (selective estrogen receptor modulator [SERM] for postmenopausal women in whom none of the medications listed above is appropriate). However, the results of multivariate random-effects network meta-analyses suggest that teriparatide and denosumab are more effective and reduce the risk of vertebral fractures more than oral bisphosphonates [60]. In patients at moderate or high risk of fracture who are being treated with anti-osteoporosis medication and discontinuing GCs, it is recommended that the treatment course should be continued until the fracture risk is reduced to a low level; then treatment should be ceased [9].
In endogenous CS, anti-osteoporotic drugs should be reserved for patients with persistent hypercortisolism not adequately corrected by specific treatments [61]. There is evidence that bisphosphonates can induce an improvement in BMD and could be useful in patients with persistent post-surgical hypercortisolism to prevent further bone loss [62]. However, it is still unknown how bisphosphonates with long-term antiresorptive effects can affect bone remodeling after successful treatment of endogenous CS. The diagnosis of CS is usually delayed, and at this stage the bone condition is usually characterized by low bone turnover, which may be further suppressed by antiresorptive drugs.
Therefore, based on the knowledge of the pathophysiology of the effect of GCs on bone, it seems that other anti-osteoporosis drugs may have a better effect in the treatment of patients with skeletal fragility caused by endogenous CS.
Denosumab has been shown to reverse the effects of GCs on bones and due to its rapidly reversible effect after discontinuation, it may be an attractive drug for short-term use in GIO caused by endogenous hypercortisolism, when curative surgery is expected [21, 63]. However, the main pathophysiological role of reduced bone formation caused by GC provides a rationale for the use of an anabolic drug such as teriparatide. Nevertheless, evidence for the safety and efficacy of teriparatide in patients with endogenous CS is still lacking.
Muscular complications of glucocorticosteroid excess
Epidemiology
Myopathy associated with GCs excess remains one of the most prominent features of CS, with the prevalence ranging from 42% to 83% [5, 64]. However, detailed examination identifies myopathy in nearly every patient with florid hypercortisolemia [6]. Interestingly, GC-induced myopathy (GIM) is slightly more common in ectopic than in adrenal hypercortisolemia [64]. Among patients with exogenous GC excess, severe muscle weakness affects 2.4% to 21% [65].
The development of GIM depends on several factors. In patients treated with exogenous GCs, the use of prednisone in doses lower than 10 mg per day (or equivalent) is usually not associated with GIM. Higher GC doses lead to a more rapid onset of clinically significant muscle weakness, which can develop within 2 weeks after the introduction of therapy with GCs [66]. The administration of 40–60 mg of prednisone per day (or equivalent) for 1 month may lead to some muscle weakness [66]. The risk of GIM is higher in patients treated with fluorinated GCs (e.g., betamethasone, dexamethasone, triamcinolone) than in patients treated with non-fluorinated GCs (e.g., prednisone and prednisolone) [66].
The way of GC administration also influences the risk of muscle damage: in the case of inhaled or epidurally injected GCs, GIM has rarely been reported [67, 68]. Other factors which may increase the risk of GIM development include older age, hyperglycemia, presence of malignancy, respiratory muscles disorder, negative nitrogen balance before the introduction of GC treatment and lack of physical activity [6, 65].
Interindividual susceptibility to GIM in endogenous CS may be attributed to various factors e.g., to GR polymorphisms. In the study of Müller et al. [69] decreased handgrip strength in hypercortisolemia was more pronounced in A3669G wild type than in A3669G minor allele carriers.
Pathophysiology
Glucocorticosteroids excess alters the structure and function of skeletal muscle in multiple mechanisms; GCs cause fast-twitch (type II) muscle fibers (mainly type IIx and IIb) atrophy with a limited or no impact on type I fibers [70, 71]. GR was proven to be essential for the development of GC-induced myopathy – the study of Watson et al. [72] showed that muscle-specific GR-knock-out mice are resistant to the effect of excess GC on muscle.
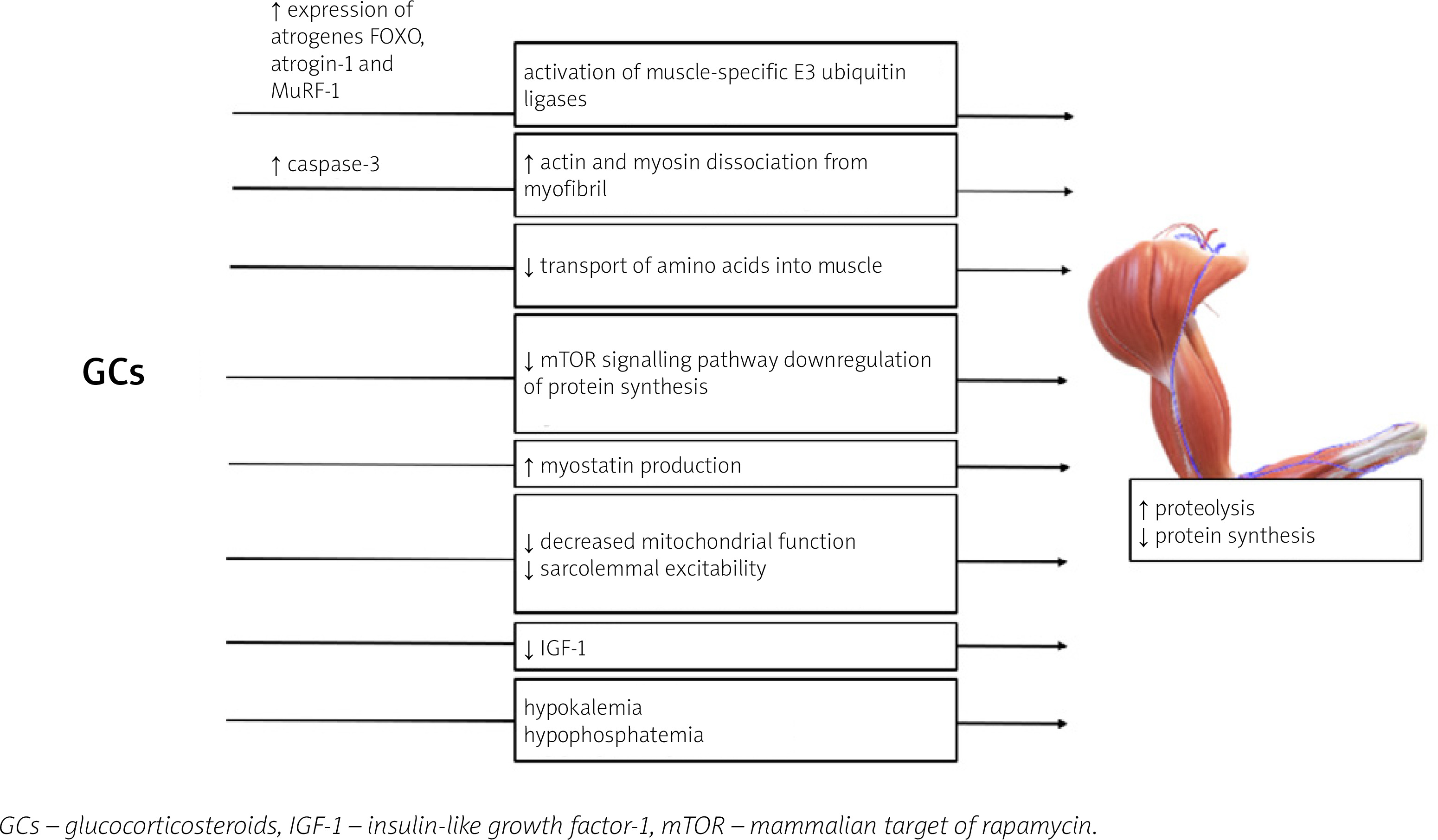
Glucocorticosteroid-induced myopathy is a consequence of increased protein catabolism and decreased synthesis [70]. Induction of muscle proteolysis occurs mainly due to the activation of ubiquitin proteasome, lysosomal systems, and calcium-mediated systems (calpains) [71]. The activation of the two former catabolic systems is regulated by the genomic actions of GC: the increased expression of numerous atrogenes (genes involved in atrophy), e.g., FOXO, atrogin-1 and MuRF-1 genes [70, 73].
The role of Forkhead box O (FOXO) transcriptional factors in the development of GC-induced muscle atrophy has been established in multiple studies [73–76]. Overexpression of FOXO leads to the activation of muscle RING-finger protein-1 (MuRF-1) and atrogin-1, which are muscle-specific E3 ubiquitin ligases, and results in increased muscle wasting [70, 76, 77]. Interestingly, the ubiquitin proteasome system is not capable of intact myofibril degradation; thus the entire process requires the activation of caspase-3 for actin and myosin dissociation [78].
The anti-anabolic effect of the excess of GC on muscles is also a result of different processes [70]. Glucocorticosteroids inhibit the transport of amino acids into muscle [70]. Furthermore, GC repress the mammalian target of rapamycin (mTOR) kinase signalling pathway, responsible for phosphorylation of eIF4E-binding protein 1 (4E-BP1) and the ribosomal protein S6 kinase 1 (S6K1), leading to downregulation of protein synthesis [79].
Glucocorticosteroids may also hinder muscle development through alterations in local growth factor production [70]. Also GCs suppress the muscle IGF-1 production [80] and IGF-1 has an ability to activate phosphoinositide 3-kinases/protein kinase and the B/mammalian target of rapamycin kinase (PI3K/Akt/mTOR) pathway, repress the transcription of FOXO, and decrease GC-induced atrophy (GIMA) [70, 81, 82].
Muscle mass development in patients with GCs excess is also affected by stimulation of myostatin (member of the transforming growth factor β [TGF-β] superfamily), production which results in decreased proliferation and differentiation of satellite cells [83]. Other postulated mechanisms leading to GIMA include decreased mitochondrial function and sarcolemmal excitability [5, 70]. Furthermore, GCs with high mineralocorticoid activity may induce hypokalemia and hypophosphatemia which may contribute to muscle weakness [65].
The mechanisms of GC excess leading to the development of GC-induced myopathy are presented in Figure 2.
Clinical presentation
Glucocorticosteroid-induced myopathy occurs in two clinical forms. Acute GIM is a rapidly progressive condition affecting proximal and distal muscle groups; respiratory muscles also can be affected [84]. This form of GC-induced myopathy is often observed in patients of intensive care units, treated with high doses of GCs [66].
Chronic GIM is characterized by painless (or mildly painful), slowly progressive proximal myopathy affecting predominantly the pelvic girdle muscles, rarely distal muscles [66]. The patients usually report difficulties while rising from a seated position, climbing stairs, and performing overhead activities [6]. Laboratory tests, including creatine phosphokinase (CPK), aspartate aminotransferase (AST), lactate dehydrogenase (LDH) and aldolase, are usually within the reference range, but mild elevation may be observed in the early stage of the disease and in the acute form of GIM [66, 85]. Muscle biopsy reveals nonspecific atrophy of type IIb fibers, no inflammatory infiltrate, rarely necrosis [66]. The electroneuromyography (EMG) result is within the normal range in the early stages, but as the disease progresses, a myopathic pattern with small-amplitude short-duration polyphasic action potentials, without spontaneous activity upon needle insertion, may be observed [66, 86]. Magnetic resonance imaging (MRI) may be helpful in differential diagnosis, to exclude inflammatory myopathies [66].
Treatment
Undeniably, in patients with endogenous hypercortisolemia and GIM, restoring the normal cortisol level through operative or pharmacological treatment is an optimal way to reverse muscle damage. In prevention and management of GIM, adequate protein intake and physical therapy (aerobic and resistance exercises) are of proven efficacy [87, 88]. Since the increased expression of IGF-1 was shown to play a protective role in GIM, it seems to be a promising therapeutic target. In an animal model, stimulation of IGF-1 and deletion of myostatin gene prevented GIM [82, 89]. Among other growth factors, ghrelin, famously known as the “hunger hormone”, produced mainly by the stomach, may also inhibit GIM [90]. Due to the hypertrophic effect on muscles, it is to be expected that androgens may also prevent GIM.
The administration of testosterone, nandrolone, selective androgen receptor modulator (SARM) and DHEA was reported to prevent the reduction of muscle strength and mass induced by GCs [91–94]. Among other agents, branched chain amino acids (BCAAs), glutamine, taurine, creatine, carbenoxolone (inhibitor of 11β-HSD1 regulating conversion of cortisone to cortisol, which decreases active GCs availability) and vitamin D have been reported to prevent GC-muscle atrophy [95–100]. However, these interventions were not conclusively evaluated in randomized controlled studies in humans and are not currently recommended. A summary of characteristics of GIO and GIM discussed in the review is presented in Table I.
Table I
Summary of features of glucocorticosteroid-induced osteoporosis and glucocorticosteroid-induced myopathy described in the review
[i] AST – aspartate aminotransferase, BMD – bone mineral density, BMP-2 – bone morphogenetic protein 2, C/EBP – CCAAT/enhancerbinding protein, CPK – creatine phosphokinase, GCs – glucocorticosteroids, GH – growth hormone, HRpQCT – high resolution peripheral quantitative computed tomography, IGF-1 – insulin-like growth factor-1, LDH – lactate dehydrogenase, mTOR – mammalian target of rapamycin, OPG – osteoprotegerin, RANKL – nuclear factor-κB ligand, TBS – trabecular bone score.
Conclusions
Musculoskeletal complications of CS contribute to the increased mortality and impaired quality of life. However, successful treatment of endogenous CS or in the case of exogenous CS discontinuation of GC therapy (if feasible) leads to improvement of the clinical course. Since the diagnosis is often delayed, it is crucial to identify patients with endogenous hypercortisolemia early and restore the normal cortisol level.
For the patients who are administered GC because of therapeutic purposes, early assessment, and introduction of pharmacological (calcium and vitamin D supplementation and bisphosphonates), behavioral (physical therapy) or dietary (adequate protein intake) interventions may help to reduce musculoskeletal complications.
Since the pathophysiology of the effect of GC excess on the musculoskeletal system is a complex interplay of different factors, to large extent unexplained, there is a need for studies providing high-quality data, which may unveil new therapeutic targets.