
Introduction
Wilson’s disease (WD) occurs due to an autosomal recessive mutation that targets the ATP7B gene on chromosome 13, which is responsible for the ATP7B protein [1]. This mutation manipulates the hepatocellular copper transportation and causes copper accumulation in the liver and other tissues such as brain and cornea [2]. Autoimmune diseases and endocrinal disorders rarely present as an associating feature of WD. Herein, we present an interesting case of WD associated with systemic lupus erythematosus (SLE), hemolytic anemia, and hypoparathyroidism.
Case report
On May 2018, a 19-year-old female patient came to our clinic complaining of slurred speech, mild difficulty with deglutition, and an abnormally hyperextended right big toe. The medical history included the previous admission at a pediatric rheumatology department on November 2015 with severe acute hemolytic anemia. At that time, the patient had a malar rash, arthralgia of both knees, oral ulcers, photosensitivity, and proteinuria of 700 mg/24 hour urine collection, for which a renal biopsy was taken and yielded a picture of thrombotic microangiopathy. Thus, the diagnosis of active SLE was attained according to the American College of Rheumatology (ACR) criteria [3].
At the primary admission, the patient’s hemoglobin was 4 mg/dl, reticulocyte count was 12 × 109/l, and lactate dehydrogenase was 1200 units/l. Coomb’s test (direct and indirect) was negative, glucose-6-phosphate dehydrogenase (G6PD) activity was normal, and the osmotic fragility test showed mild resistance to hemolysis. The autoimmune profile, including antinuclear antibody (ANA), anti-double-stranded DNA antibody (anti-dsDNA), anti-Smith antibodies, and antiphospholipid antibodies (APLAs), was negative. The liver enzymes (aspartate transaminase (AST) and alanine transaminase (ALT)) were elevated by 2–3-fold at different follow-up points. Ultrasound abdomen and pelvis showed mild hepatomegaly. Autoimmune hepatitis was excluded by negative smooth muscle antibody (SMA) and liver kidney microsomal type 1 (anti-LKM-1) antibodies. The patient was diagnosed with SLE and started pulse steroid then maintenance steroid and hydroxychloroquine after improvement.
On the second admission (May 2018), the patient reported improvement of joint pain and oral ulcers on steroid therapy. Upon examination, the patient was stable with dysarthria, tremors, evident malar rash, and right hyperextended big toe. There was no sensory or motor function loss. The ANA, anti-dsDNA, and APLAs were immediately ordered. Upon neurological consultation, brain magnetic resonance imaging (MRI) and electroencephalography (EEG) were recommended. The patient started pulse steroid and was planned for cyclophosphamide therapy. The ANA was positive, anti-dsDNA was equivocal, and APLAs were negative. The EEG was normal, but the brain MRI showed bilateral symmetrical basal ganglia abnormal signal intensity.
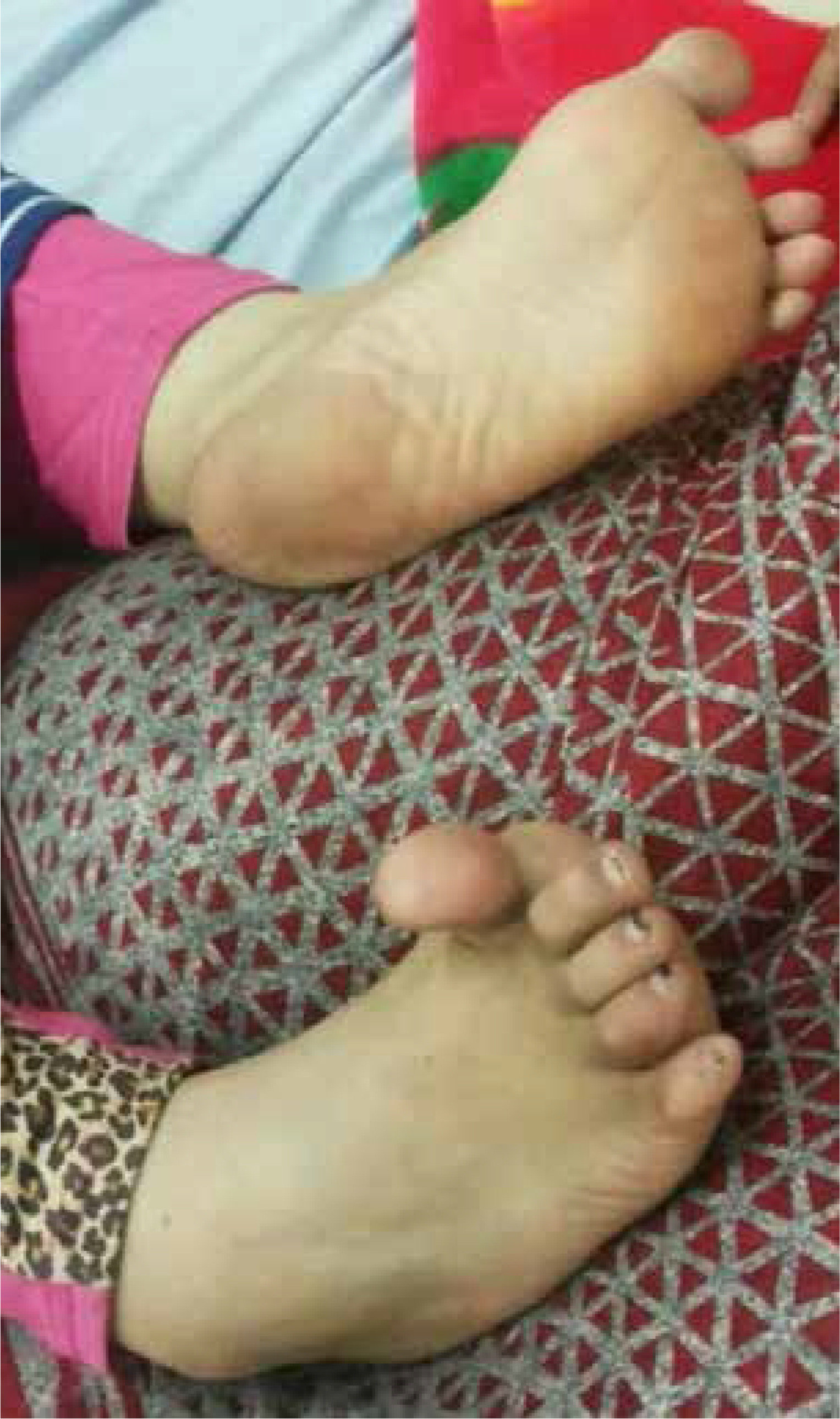
The neurologists suspected the presence of vasculitis or neuroinflammation, but they recommended investigating the patient for metabolic causes as this symmetrical involvement of the basal ganglia is rare in lupus vasculitis and may have an underlying metabolic cause. Hypoparathyroidism was detected (ionized calcium was low normal 1.1 mmol/l, phosphorus was 3.9 mg/dl, and parathyroid hormone (PTH) equaled 9 pg/ml). Ophthalmological consultation confirmed the presence of Kayser-Fleischer ring. The serum ceruloplasmin was 3.1 mg/dl, and 24-hour urinary copper was 1568 μg/day. The diagnosis of WD was made, and the patient started receiving oral zinc 150 mg/day. By that time, the extrapyramidal signs extended to the left foot with a similarly hyperextended big toe (Fig. 1). Significant improvement in speech and deglutition was noted within one month of receiving oral zinc acetate.
Discussion
Wilson’s disease is an inherited metabolic abnormality that causes accumulation of copper in various organs, mainly the liver, brain, and cornea [1, 2]. Hepatic involvement is the most common presenting feature followed by neurological and psychiatric manifestations [4].
The patient first presented with unexplained acute severe hemolytic anemia at 16 years old. The diagnosis of WD had not yet been reached at this stage, and the patient had typical features of SLE, so the anemia was attributed to SLE. This is consistent with previous cases in the literature where the occurrence of hemolytic anemia was reported before detecting WD. For instance, Ye et al. [5] reported a 15-year old girl who had acute hemolytic anemia as the first presentation of WD. However, the diagnosis was not reached until recurrence of the hemolytic attack with hepatic and neurological deterioration. Similarly, Santra et al. [6] reported two cases of acute hemolytic anemia of unclear etiology as an initial presentation of WD.
At the first admission, the patient was diagnosed with SLE based on clinical features. The association between SLE and WD is rarely reported in the literature and is not yet clearly understood. Santhakumar et al. [7] reported a 24-year old female patient who was first diagnosed with SLE and secondarily with antiphospholipid syndrome. The diagnosis of WD accidentally occurred when they sent the patient for ophthalmological evaluation due to the diminution of vision. The Kayser-Fleischer ring was detected, and low serum ceruloplasmin concentration and high 24-hour urinary copper confirmed the diagnosis [7].
More recently, Zhang et al. [8] described a case of a young woman who was diagnosed with SLE and WD at the same time. She presented with typical neuropsychiatric symptoms of involuntary limb movement and dysarthria. The slit lamp examination, low serum ceruloplasmin, and high 24-hour urinary copper confirmed the diagnosis. At the same time, the patient had manifestations and positive serology for SLE.
At the second admission, the MRI findings together with the hand tremors urged assessment of the ionized calcium, phosphorus, and PTH, which indicated the presence of hypoparathyroidism. Endocrinal disorders are rarely reported in WD, and only a few cases have described the association of hypoparathyroidism and severe refractory hypocalcemia with WD.
In 1983, Carpenter et al. [9] reported the case of an 11-year old girl with WD who presented with hypoparathyroidism, which was attributed to deposition of copper in the parathyroid glands. Another recent report described a case of a 16-year old boy who presented with tetany with dystonia of the limbs and orofacial region. Upon investigation, the patient had hypoparathyroidism. Later on, he experienced extrapyramidal symptoms that assisted in the diagnosis of WD [10]. The patient’s big toe involuntarily dorsiflexed was not a typical positive Babinski sign. There was no fanning out of other toes. The dorsiflexion was voluntarily reversible. There is a similar condition in the literature known as painless legs moving toes syndrome [11].
The patient fulfilled more than four out of eleven items according to the ACR criteria for the diagnosis of SLE [3]. In addition, a renal biopsy revealing thrombotic microangiopathy was confirmed by the pediatric rheumatology department, which is thought to be secondary to SLE. It is also worth mentioning that our patient had a significant improvement of arthritis, photosensitivity, and oral ulcers on low dose steroid and hydroxychloroquine. While acute hemolysis is not an uncommon feature in the SLE setting, it is, however, a rare presenting feature of copper overload in WD [12, 13]. Similarly, the neuropsychiatric disturbance is not a rare finding in both conditions; lupus cerebritis and thrombotic thrombocytopenic purpura (TTP) in SLE patients and symmetrical basal ganglia involvement with extrapyramidal manifestations in WD patients [14, 15].
Our patient had elevated liver enzymes and autoimmune hepatitis was suspected since previous reports revealed a common association between SLE and this immune marke [16]. However, all autoimmune markers were negative. At the second admission, abdominal US revealed cirrhotic liver that was mostly attributed to copper overload. Eventually, Kayser-Fleischer ring together with reduced serum ceruloplasmin and elevated 24-hour urinary copper strongly suggested that our patient had a rare association of SLE and Wilson’s disease. Similarities between symptom of both diseases (Wilson’s disease/SLE) are shown in Table I.
Table I
Comparison of symptoms of Wilson’s disease and systemic lupus erythematosus (SLE)
The usefulness of zinc in the maintenance treatment of copper accumulation disease has been confirmed, and this treatment was approved by the FDA [17]. However, zinc is not included in the recommendations as first-line treatment in WD such as penicillamine and triethylenetetramine (Trientine) [18]. In the case of the described patient, a decision was made about zinc use in therapy in the absence of penicillamine and Trientine on the pharmaceutical market in Egypt. Zinc effectiveness depends on blockage of intestinal copper absorption by induction of metallothionein in enterocytes and hepatocytes, a blockade of copper reabsorption and also inhibition of endogenous zinc secretion in e.g. saliva and gastric juice [19]. The described patient due to SLE was treated with methylprednisolone and hydroxychloroquine. This combination of drugs has shown its effectiveness.
Conclusions
To sum up, WD has diverse presenting features and reports of associated rare manifestations are increasing. SLE may be associated with WD, and presence of neurological, behavioral, or liver function abnormalities should raise the suspicion, even without apparent features of WD. We also encourage physicians to look for WD in patients with hemolytic anemia of unknown etiology, especially with elevated liver enzymes. Furthermore, although it is rare, we may encounter the existence of unexplained endocrinal disorders such as hypoparathyroidism as a presenting symptom of WD.