
Introduction
First described in 2008, H syndrome (HS) [ORPHA: 168569; OMIM #612391] is an uncommon autosomal recessive genodermatosis belonging to the histiocytosis-lymphadenopathy plus syndrome spectrum [OMIM: 602782]. Approximately 100 globally reported cases exist to date [1], encompassing affected individuals of North African, Arab, and Indian origins with consanguineous parentage.
Pathognomonic skin findings, including hyperpigmentation, hypertrichosis, and sclerodermatous induration, serve as the hallmark of the spectrum. However, HS further presents with the infiltration of dysregulated histiocytes, resulting in multisystem impairment.
The pathogenesis stems from loss-of-function mutations within the SLC29A3 gene (chromosome 10q23) encoding the human equilibrative nucleoside transporter 3 (hENT3) [2, 3]. As an intracellular transporter of nucleosides, hENT3 plays an essential role in facilitating the uptake of precursors for nucleotide synthesis and salvage across the lysosomal and mitochondrial membranes [4–6]. Disruption of hENT3 leads to defective histiocyte apoptosis, unchecked proliferation, and multi-organ infiltration [7, 8].
Among the initial 79 reported cases, documented musculoskeletal manifestations include flexion contractures (56%) and hallux valgus deformities (30%). Hearing loss, heart anomalies, hepatosplenomegaly, hypogonadism, hyperglycaemia (insulin-dependent diabetic mellitus), low height, and haematological abnormalities also commonly arise in patients with this disorder, hence the designation “H” syndrome [9].
This report describes the case of a 24-year-old patient presenting with characteristic dermatological findings and valgus deformities. Their evaluation ultimately confirmed a diagnosis of HS based on clinical and histological findings.
Methods
A comprehensive narrative review was conducted to address the question: “What is H syndrome, and what are the rheumatological manifestations of this rare syndrome?” Initially, we performed a broad search in PubMed using the terms “H syndrome SLC29A3”, which yielded 76 results as of March 2024. To ensure comprehensive coverage of this rare condition, no date restrictions were applied.
These 76 results were screened for relevance and categorised. Articles were included if they focused on HS or compared it to other SLC29A3-related disorders, described clinical manifestations (especially rheumatological features), discussed epidemiology, underlying mechanisms, or treatment options. Case reports, case series, review articles, and original research papers in English were considered.
We omitted several articles that mainly focused on HS genetic mutations, on specific organ manifestations, primarily discussed other SLC29A3-related disorders, or were preliminary case reports lacking formal classification.
Following this initial screening, the reference lists of selected key articles were examined to identify additional relevant publications not captured in the initial database search. This citation searching process allowed us to include important studies that may have been missed in the original PubMed search.
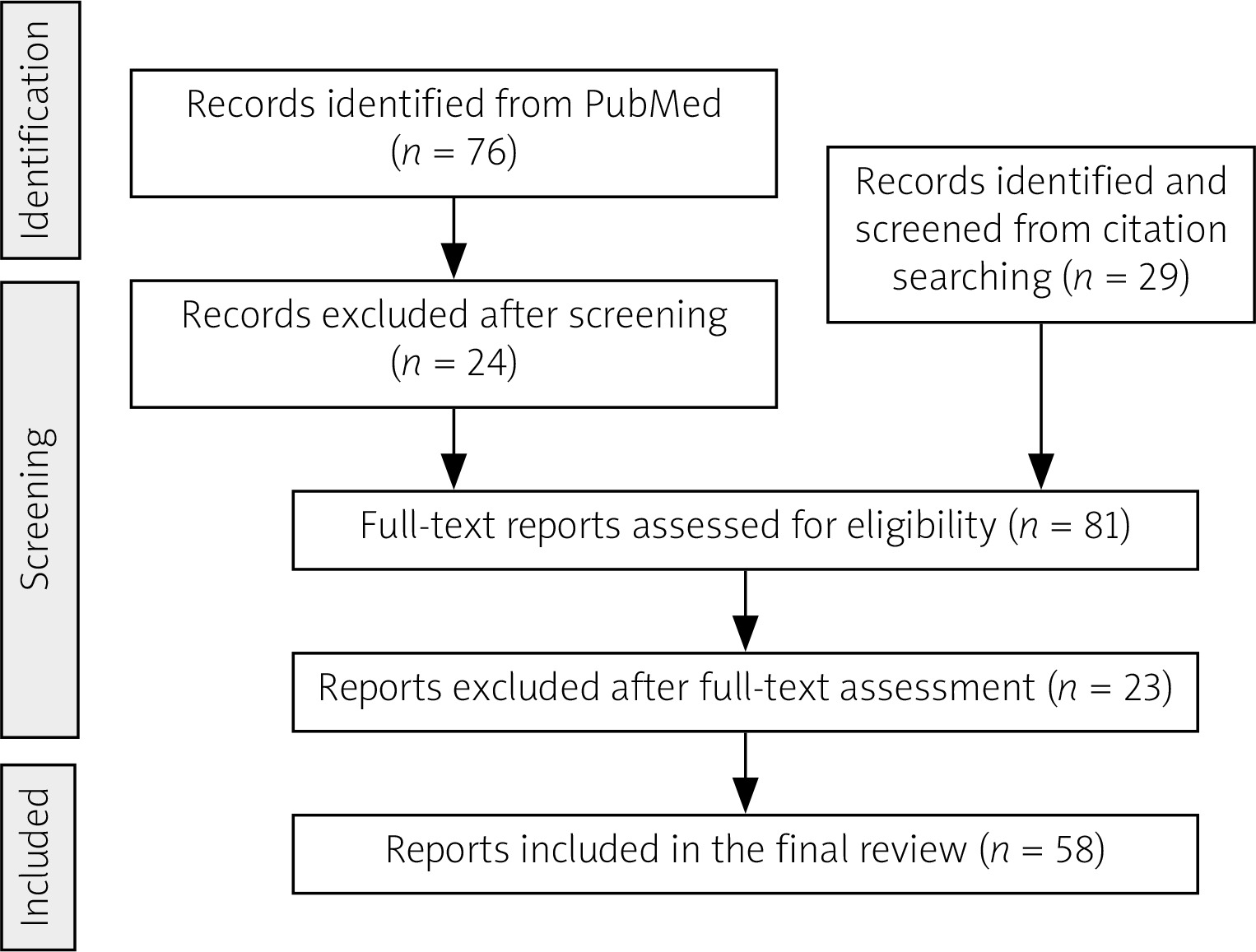
After careful assessment based on established selection parameters, 58 articles were deemed eligible and included in the final review, one of which was not indexed on PubMed. These included 48 case reports and studies, 4 case-based literature reviews, one literature review, and 5 experimental studies (Fig. 1).
Fig. 1
Flow diagram of the article selection process. The process included initial database searching (PubMed), citation tracking, and application of inclusion/exclusion criteria. Included articles focused on H syndrome’s clinical manifestations (particularly rheumatological features), epidemiology, mechanisms, and treatments. Excluded were articles primarily on genetic mutations, single-organ involvement, other SLC29A3 diseases, or unlabelled early case descriptions.
Case description
We present the case of a 24-year-old Moroccan male with HS. The patient was initially diagnosed with insulin-dependent diabetes mellitus at age 8. No family history of similar conditions was reported.
At the age of 14 years, the patient started displaying well-defined, symmetrical, non-pruritic, and painless hyperpigmented cutaneous patches with overlying hypertrichosis and induration. These patches first appeared on the lower limbs, inner thighs, groin area, genitals, lower back, and chest.
Antinuclear antibody (ANA) testing turned positive (IIF, using HEp-2 substrates; 1:80, speckled pattern), and skin biopsy revealed a chronic inflammatory perivascular infiltrate of lymphocytes and plasma cells with no histological evidence of autoimmune disease. Initial treatment with topical glucocorticosteroids (GCs) and cold cream yielded no clinical improvement.
Subsequently, at 17 years old, the patient exhibited bilateral conjunctival hyperaemia, accompanied by dilated lateral scleral vessels and a corneal arcus revealed by ophthalmic examination. Brain scan, however, showed bilateral exophthalmos. Upon clinical findings, lymph node and abdominal ultrasounds were demanded, revealing multiple axillary and bilateral inguinal adenopathy with homogeneous hepatomegaly, respectively. Trans-thoracic echocardiography was normal. Plasma protein electrophoresis detected polyclonal hypergammaglobulinaemia.
Hormonal evaluation demonstrated elevated luteinizing hormone (LH) levels of 14.70 mUI/ml (1.2–10), follicle-stimulating hormone and prolactin levels were within normal ranges at 12.35 mUI/ml (1.5–14) and 10.60 ng/ml (5–15), respectively.
At the age of 23 years, diabetic retinopathy was confirmed. Scrotal ultrasound showed bilateral testicular hypotrophy with microlithiasis. Audiometry revealed moderate hearing loss, predominantly on high-pitched sounds.
In December 2023, the patient was addressed to the rheumatology department for an evaluation of painful swelling of the feet, present since age 12 and exacerbated by walking, particularly in the posterior heel.
Osteo-articular examination revealed notable ankle and foot swelling. Passive dorsiflexion was measured at 20 degrees, whereas active dorsiflexion was restricted, reaching only 15 degrees. Extension reached 30 degrees with passive mobilisation and 20 degrees through active mobilisation. Additionally, the patient displayed a pes planus with a fixed toe claw during plantar flexion, along with a noticeable hallux valgus (Fig. 2).
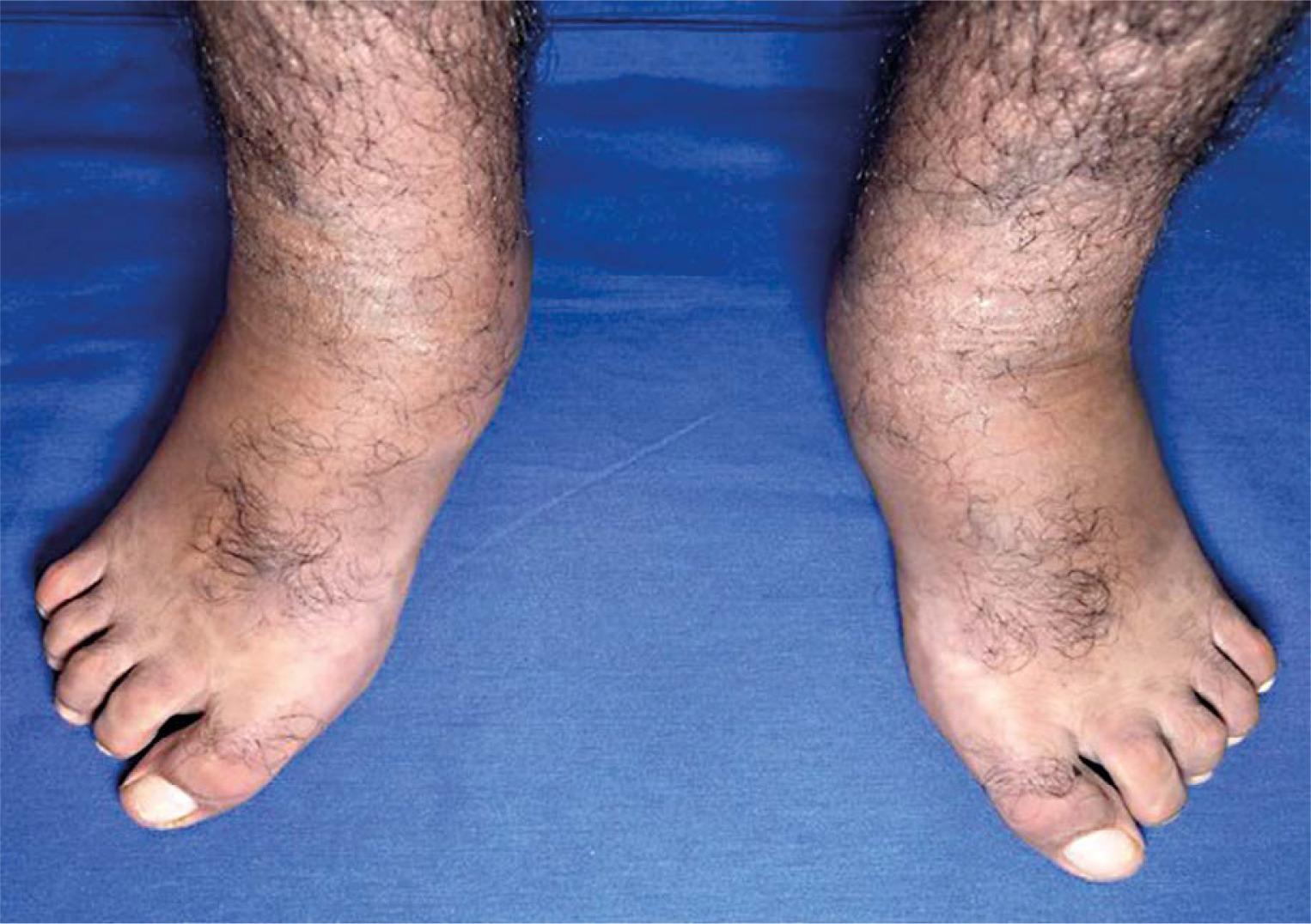
Further physical examination revealed no apparent clinical signs of synovitis, arthritis, or joint restrictions in the upper limbs, knees, or hips. Axial assessment demonstrated a flexible spine, devoid of pain or discomfort. Despite the presence of an externally rotated stance of the feet, the patient was able to walk without assistance. Furthermore, standing balance was preserved, and neurological evaluation yielded normal findings.
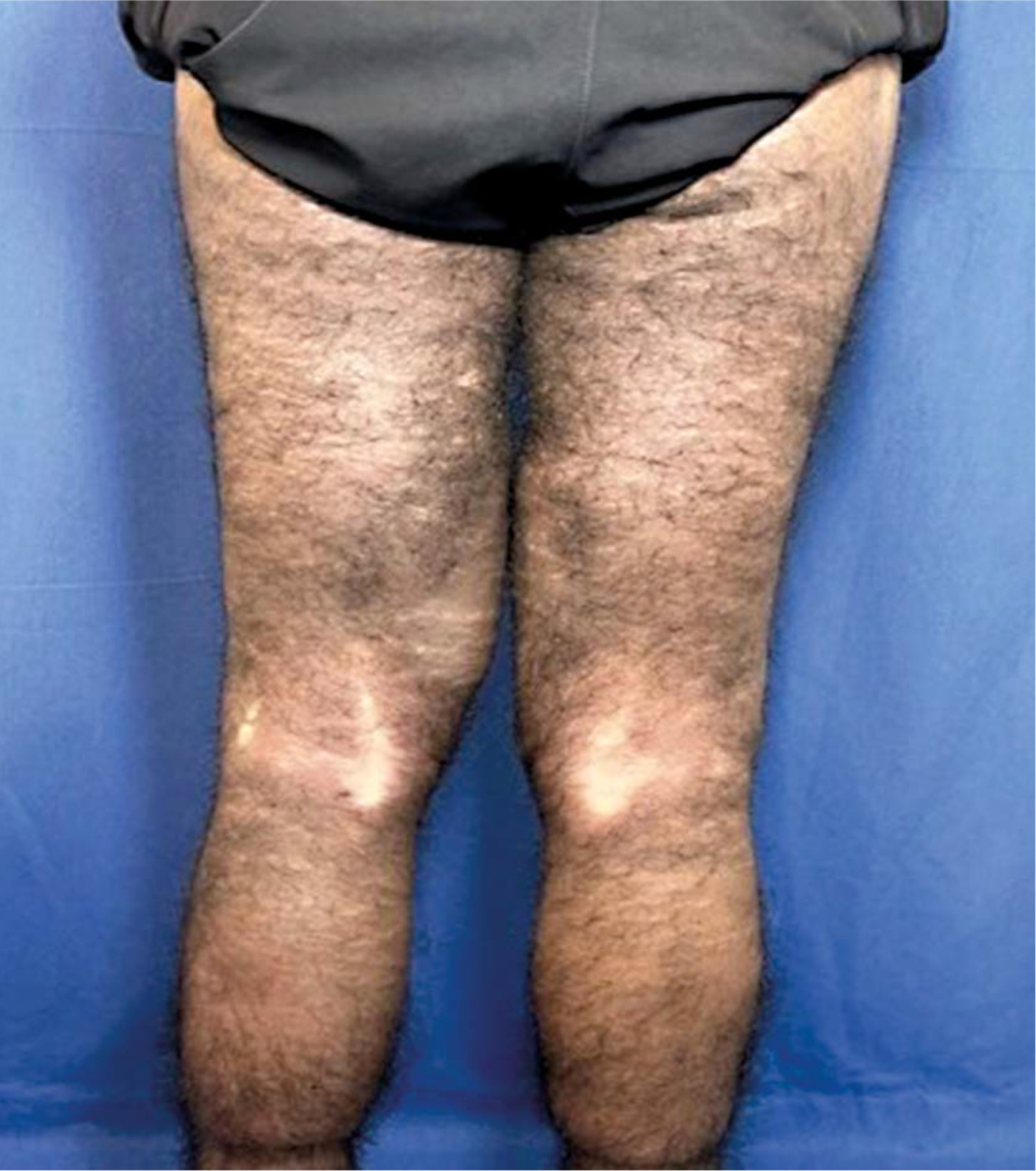
Skin examination indicated progression of cutaneous patches to the upper limbs, trunk, and abdomen, sparing the knees, popliteal fossa, and medial aspect of the buttocks (Figs. 3–5).
Fig. 3
Hyperpigmented cutaneous patches with hypertrichosis and induration on lower limbs and sparing the knees (anterior view).
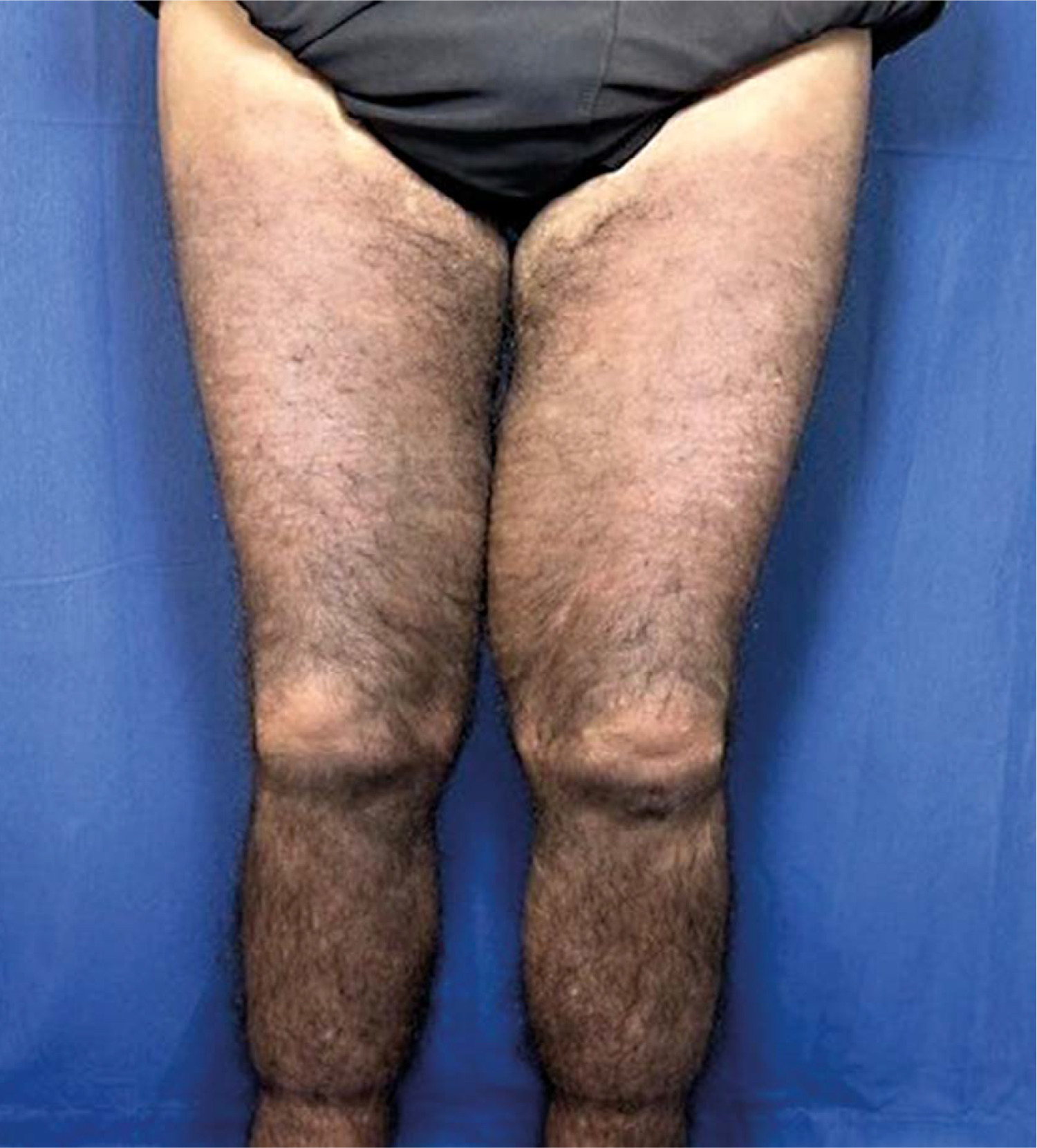
Fig. 4
Hyperpigmented cutaneous patches with hypertrichosis and induration on lower limbs and sparing the knees (posterior view).
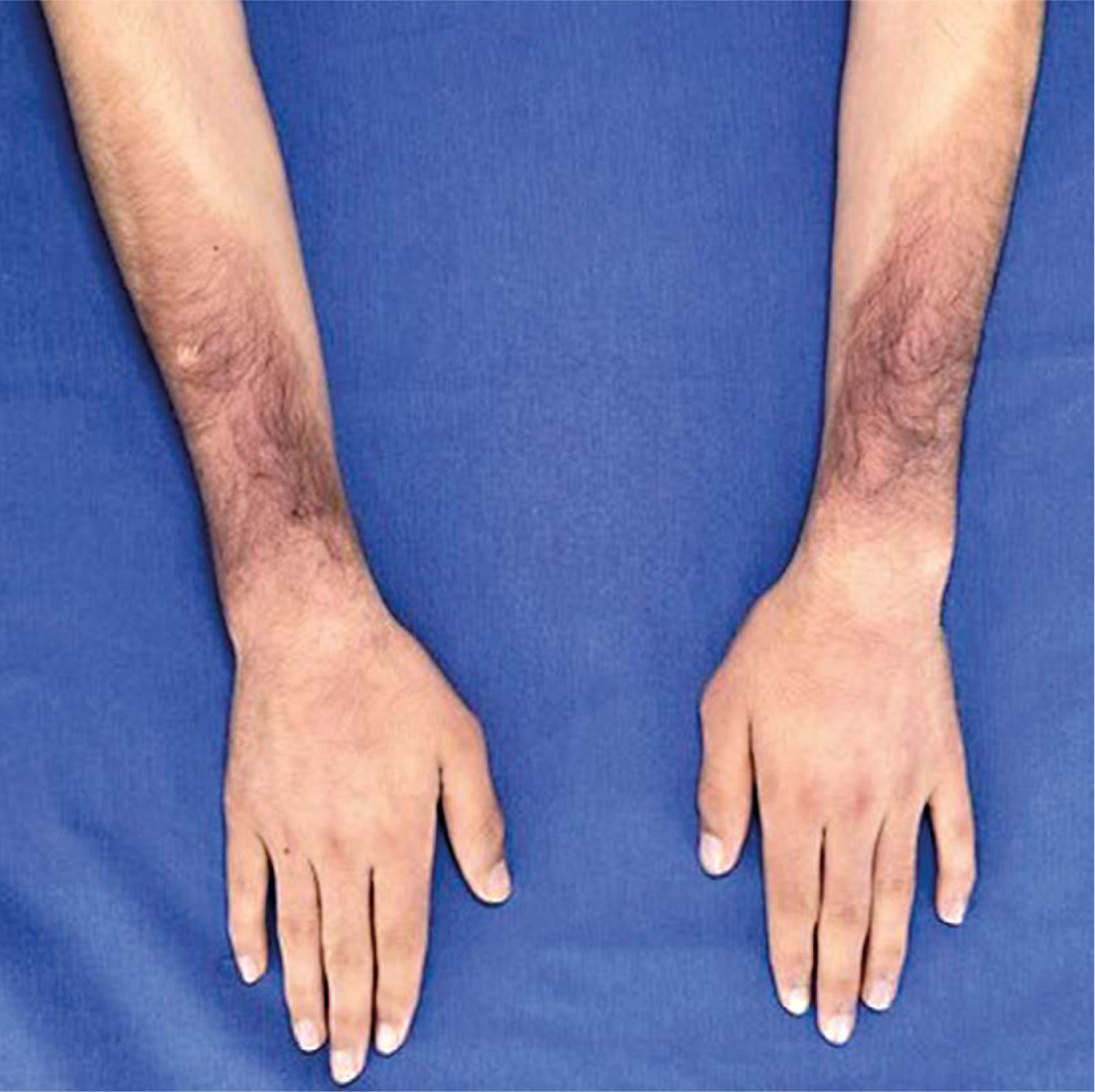
Laboratory investigations revealed several abnormalities: elevated glycated haemoglobin 12.2%, along with fasting glucose level (1.11 g/l) (0.70–1.10), erythrocyte sedimentation rate (ESR) 66 mm/h (< 10.5), and C-reactive protein (CRP) 3.60 mg/l (< 5). Blood count showed anaemia at 12.1 g/dl. High-density lipoprotein (HDL) cholesterol was lower than the reference range at 0.25 g/l (0.40–0.60) and triglycerides at 1.60 g/l (0.3–1.5). In contrast, liver, renal, and thyroid function test results were unremarkable.
Radiographs of the feet in anteroposterior, lateral, and three-quarters views did not show arthritis, heel spurs, or joint destruction. However, an X-ray of the lumbar spine indicated a sacralisation of L5. While dual-energy X-ray absorptiometry (DEXA) revealed lumbar osteopaenia (T-score = –1.1). X-ray of the pelvis showed no sacroiliitis.
Ankle ultrasound revealed bilateral calcaneal enthesopathy characterised by irregularity, erosion, and calcification. There was no evidence of arthritis in the talocrural, talonavicular, or subtalar joints. The extensor and flexor tendons of the toes, as well as the fibular tendons, exhibited normal conditions. Moreover, there was extensive infiltration observed in the subcutaneous tissue.
The thoraco-abdomino-pelvic computed tomography (CT) scan showed homogeneous hepatomegaly, as well as a malformation of the vena cava return. The CT scan also revealed bilateral inguinal adenopathy and a normal-sized spleen.
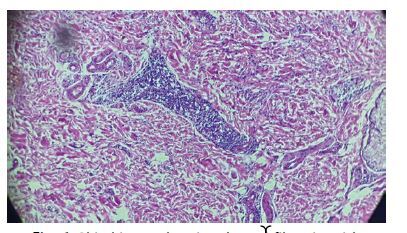
On the skin biopsy, a normal epithelium with orthokeratotic stratum corneum was found overlying a dermis composed of fibrous tissue. The dermis contained a moderate to significant inflammatory infiltrate consisting of mature lymphocytes forming cuffs and macrophage histiocytes with foamy cytoplasm, distributed around large blood vessels, hair follicles, and within the interstitium (Fig. 6). Hair follicles were sparsely represented and showed focal follicular mucinosis.
Fig. 6
Skin biopsy showing dermal fibrosis, with perivascular lymphohistiocytic inflammatory infiltrate.

The final diagnosis of HS was established based on the characteristic clinical and histopathological findings.
Discussion
Mechanism
Mutations in the SLC29A3 gene underlie HS as well as other diseases related to its dysfunction [2, 10, 11]. This gene encodes the pH-dependent hENT3.
Part of the ENT family – a type of solute carrier (SLC) transporter, hENT3 regulates intracellular nucleoside trafficking from the lysosomes to the cytoplasm [4] and across the inner mitochondrial membrane [5]. This widely distributed transporter is essential for nucleotide salvage, particularly in haematopoietic cells lacking de novo nucleotide synthesis. The nucleic acids transported by hENT3 originate from apoptotic cells that have been phagocytosed by macrophages and histiocytes.
Consequently, nonfunctional hENT3 in HS – typically by a loss-of-function mutation – results in accumulation of undigested materials in macrophage lysosomes, disrupting their integrity and functions and impairing macrophages’ ability to clear apoptotic cells. This impairment triggers macrophage-colony stimulating factor/macrophage-colony stimulating factor receptor signalling, which drives macrophage proliferation [7].
Furthermore, the development of histiocytosis in HS has also recently been attributed to the stimulation of toll-like receptors (TLR), namely TLR7 and TLR8, expressed in the lysosomal membrane of monocytes. These receptors are persistently activated by the accumulation of nucleosides in lysosomes, subsequently upregulating the MAPK pathway, a signalling commonly enhanced in other forms of histiocytosis, including Langerhans cell histiocytosis, due to somatic mutations. Consequently, an overexpression of inflammatory genes occurs, leading to inflammation and enhanced cytokine release, and ultimately driving the formation of histiocytosis [12].
Epidemiological profile and clinical findings
H syndrome typically manifests during the first or second decade of life [13]. However, cases of HS diagnosed at an advanced age have been reported, including a family with 2 brothers whose median age at diagnosis was 53 years, and their 51-year-old female cousin, described by Chouk et al. [14]. Our patient’s age at onset aligns with the typical range reported in the literature.
Initially reported in individuals of Arab ethnicity by Molho et al. [2, 9, 13], cases of HS have since been documented across diverse Arab countries (Egypt [15, 16], Iraq [17, 18], Syria [19], and Saudi Arabia [20]) and ethnic backgrounds worldwide, including North Africa (e.g. Tunisian [14, 21, 22] and Algerian [23] patients), India [24–26], and other regions, i.e. the United States [27], Turkey [28], Iran [29] Soudan [30], and China [31, 32]. This global prevalence highlights that while the syndrome was first described in a particular population, it is not confined to any specific ethnic group.
To date, few cases have been described in the Moroccan population [2, 9, 33–39].
H syndrome exhibits a highly heterogeneous clinical phenotype, characterised by infiltration of histiocytes and multisystemic dysfunction. Manifestations encompass mucocutaneous features such as hyperpigmented and indurated skin lesions, hypertrichosis, as well as systemic involvement including hepatomegaly, cardiovascular abnormalities, sensorineural hearing loss, hyperglycaemia, hypogonadism, flexion contractures, and short stature [9, 13].
These clinical manifestations are shared with 3 other disorders within the SLC29A3 spectrum, previously considered distinct entities: Faisalabad histiocytosis [11, 15, 21, 35, 40], sinus histiocytosis with massive lymphadenopathy[11, 14, 21], and pigmented histiocytosis with insulin-dependent diabetes [10, 15, 21, 35].
Among the diverse manifestations, the skin features are particularly prominent and prevalent, observed in approximately 68% of affected individuals. They are characterised by hyperpigmentation, hypertrichosis, and induration [9]. These thickened, hyperpigmented plaques typically present initially on the inner thighs and gradually extend to the lower and middle abdominal regions, genitalia, shins, and feet. Notably, the knees and buttocks are often spared, a feature considered distinctive of HS [2]. In our case, the patient’s cutaneous manifestations align with the classical mucocutaneous distribution pattern described in the literature.
Musculoskeletal and osteoarticular manifestations
The 3 most commonly reported rheumatological manifestations in the first 79 documented cases of HS are flexion contractures (56%), hallux valgus (30%), and other foot deformities (20%) [9].
Fixed flexion contractures have been observed affecting the proximal interphalangeal (PIP) joints, while the distal interphalangeal joints exhibit hyperextension deformities, simulating a boutonniere deformity. True dislocation and erosion of the PIP joints have also been described in 6 patients out of 12 [41]. Similar deformities involving the toes, without true dislocation, were observed in our patient.
Hallux valgus deformity is generally reported as bilateral [15, 18, 28], although it may manifest unilaterally [42], and may be accompanied by pes planus [9, 27, 39] and heel valgus [43].
Other deformities have been described such as lateral tibial torsion, genu valgum, and cubitus valgus [2, 25, 44].
Moreover, several articles refer to arthritis, with or without erosive features. It is increasingly prevalent among younger patients [8, 45], contributing to the misdiagnosis of HS as juvenile idiopathic arthritis [18, 20, 42, 46].
One exceptional case involved axial inflammatory conditions, specifically bilateral sacroiliitis [46].
Bone damage includes osteopaenia, osteoporosis, and consequently bone fractures [9, 24]. Reduced bone mineral density may be attributed to untreated hypovitaminosis D present in these patients, which not only increases the risk of fracture, but also aggravates bone deformities [17].
Widening of the bones, predominantly affecting the hands [41] and long bones (femora), is characterised by metaphyseal flaring accompanied by intramedullary infiltration [21] in addition to cortical hyperostosis and thickening of the calvaria.
However, enthesis involvement of the calcaneal tendon found in this patient represents a novel finding not previously documented in the literature.
Other systemic manifestations
The patient presented with clinical manifestations consistent with the literature on the HS, including hepatomegaly, hearing loss, inguinal lymphadenopathy, insulin-dependent diabetes mellitus, primary hypogonadism manifested by micropenis, dilated scleral vessels, and exophthalmos. However, certain phenotypes were notably absent.
Although short stature is prevalent in approximately 49% of HS cases [9], our patient displayed normal adult height of 174 cm. Cardiovascular abnormalities, predominantly pericardial lesions occurring in around 34% of cases, were not detected as comprehensive cardiovascular evaluation, including transthoracic echocardiography, showed unremarkable results. Sensorineural hearing loss, a frequent feature affecting 53% of SLC29A3 mutants [9], was also present in this case. Other clinical features documented in the literature but absent in this case included splenomegaly (39%), genital masses (29%), varicose veins (28%), facial telangiectasia (27%), gastrointestinal involvement (15%), renal abnormalities (6%), fever (5%), intellectual disability (5%), gingival hypertrophy (3%), and swollen cheeks (1%) [9].
Multiple reports have described the variable clinical manifestations of HS ranging from mono- or paucisymptomatic forms [8, 34] to combined phenotypes of the SLC23A9 spectrum diseases [15, 16, 35], highlighting the complexity and diverse manifestations of this rare disorder.
Despite the genetic basis and shared histiocytic features, the clinical presentation can vary significantly among individuals, even within the same family demonstrating phenotypic intrafamilial variability and a lack of genotype-phenotype correlation [8, 9, 28]. This heterogeneity poses challenges in diagnosis and management, underscoring the importance of comprehensive clinical evaluation and consideration of the entire SLC29A3 spectrum of diseases.
Biology
This case highlights several cardinal features of HS that are consistently reported across the literature. Foremost, the inflammatory state manifested by elevated ESR and CRP levels aligns with the well-established inflammatory nature of this disorder [9, 13, 28]. The driving forces behind this dysregulated inflammatory response is suggestibly rooted in the persistent activation of MAPK-TLR signalling, due to the accumulation of nucleosides inside monocytes’ lysosomes, leading to cytokine secretion [12]. This chronic inflammation may lead to organ damage. Thus, inflammatory profiling in patients with HS is recommended, for evaluating pretherapeutic and post-therapeutic state [47].
The microcytic anaemia observed in our patient is observed in 23% of patients with HS [9], frequently attributed to chronic inflammation with normal to high ferritin levels and low iron binding capacity [13, 20, 28] though it can also be related to iron deficiency [14]. Furthermore, cases of autoimmune haemolytic anaemia have been reported with a positive direct Coombs test [48–50] testifying the autoimmune aspect of this disorder’s manifestations [28].
The detection of polyclonal hypergammaglobulinaemia on plasma protein electrophoresis is a notable finding frequently documented across various case series and reports [14, 23, 28, 34, 45, 51, 52]. This benign condition probably reflects the dysregulation of the immune system in HS, which involves polyclonal activation of B-cell and plasma cell populations in the context of systemic inflammation [28].
Presented patient’s history of positive ANA with a speckled pattern is an intriguing observation because autoantibody positivity is not a universally reported feature in HS. Previous case reports described a similar finding [27, 52, 54].
Vitamin D deficiency and the subsequent development of hyperparathyroidism could potentially arise from various factors, including malabsorption, chronic inflammatory conditions, and insufficient exposure to sunlight. In patients with HS, low vitamin D may be associated with decreased height, bone abnormalities, musculoskeletal pain, and reduced bone mineral density. If not addressed, this deficiency could lead to the exacerbation of bone deformities and fractures [17].
The endocrine manifestations observed in this case, namely hypogonadism and insulin-dependent diabetes mellitus (IDDM), are well-established features of HS [9, 13, 15, 27, 34, 35, 39, 46, 49, 51, 53]. The elevated luteinising hormone level, in conjunction with the reported variability in gonadotropin levels (hypergonadotropic [49, 50, 53] or hypogonadotropic [15, 16, 28, 51]), highlights the complex and multifaceted nature of the endocrine dysfunction in this disorder. Similarly, the elevated glycated haemoglobin and fasting glucose levels are consistent with the strong association between HS and IDDM. Interestingly, while many reported cases of HS have documented abnormalities in liver function, thyroid function, or both [13, 18, 25, 27, 37, 53], our patient’s liver, renal, and thyroid function tests were within normal limits.
The dyslipidaemia observed in our patient, characterised by low HDL cholesterol and slightly elevated triglyceride levels, is a noteworthy finding that has not been extensively explored in the context of HS. Although hypertriglyceridaemia has been previously linked to this disorder [9, 15, 25, 48], the specific pattern of lipid abnormalities observed in this case may represent a distinct metabolic disturbance.
Histology
The histopathological findings of HS, a recently defined genodermatosis, exhibit distinct features that can vary in presentation. Typically, the epidermis displays hyperkeratosis and acanthosis, indicative of abnormal keratinocyte proliferation and differentiation [55] with an increased number of melanocytes in the basal layer, contributing to the characteristic hyperpigmentation observed in this condition [13, 15, 28, 55]. However, in our case, the skin biopsy revealed a normal epithelium with orthokeratotic stratum corneum.
The dermal and subcutaneous layers usually demonstrate marked fibrosis and infiltration of CD68-positive histiocytes, suggesting a chronic inflammatory process [13, 28, 55]. In this instance, the dermis was composed of fibrous tissue, containing a moderate to significant inflammatory infiltrate. This infiltrate consisted of mature lymphocytes forming cuffs and macrophage histiocytes with foamy cytoplasm, distributed around large blood vessels, hair follicles, and within the interstitium, which aligns with the typical perivascular and interstitial infiltrate of lymphocytes described in literature [13, 15, 28, 55]. While literature reports often note thickened collagen bundles in the dermis, this feature was not prominent in the current case. Additionally, hair follicles were scarcely represented, a finding not typically emphasised in standard descriptions of HS.
Immunohistochemical analysis has revealed that the lymphoid population comprises predominantly T cells (CD3-positive), with a minor component of B cells (CD20-positive) [13]. The histiocytic cells infiltrating the skin exhibit immunophenotypic features consistent with emperipolesis, expressing CD34, CD68, CD163, and S-100 protein, resembling the findings observed in Rosai-Dorfman disease [28, 56]. Emperipolesis is a phenomenon characterised by the presence of one cell within the cytoplasm of another, commonly found in Rosai-Dorfman disease [56].
Differential diagnosis
In this patient’s case, the diagnosis of HS was made based on the clinical presentation and histological findings. Genetic testing would be the ideal way to confirm the diagnosis; however, it was not performed due to a lack of technical resources in our setting. Therefore, we must carefully consider other potential diagnoses that cannot be definitively ruled out.
The skin lesions seen in HS are prone to being misdiagnosed as morphea profunda [8, 27, 36, 42, 54], which probably occurred with our patient during childhood – a pattern seen in many early reported cases. Both conditions feature thickening of the subcutaneous tissue and fascia, inflammatory infiltrates, collagen deposition in the dermis leading to fibrosis, and hyperpigmentation over the lower abdomen, limbs, and trunk. A key difference is that morphea profunda lacks the hypertrichosis and systemic involvement characteristic of HS. Additionally, the inflammatory infiltrate in morphea shows a predominance of plasma cells and lymphocytes rather than the histiocytes typical of HS [48]. Pigmented brown lesions, associated with diabetes mellitus or hyperglycaemia, could also raise suspicion for acanthosis nigricans. In a paediatric patient, arthritis, added to short stature and low insulin-like growth factor-1 levels, might suggest juvenile idiopathic arthritis [16, 27, 39, 42, 46], while in adults, rheumatoid arthritis would be a consideration [51]. Finally, the fixed flexion contractures of the toes and fingers could mimic an inflammatory deforming arthropathy [42].
Treatment
There is no consensus on a standardised treatment approach for HS due to the complexity of its pathophysiology. Multiple treatments have been attempted, with no or partial therapeutic success, including anti-inflammatory agents such as the following: systemic GCs, nonsteroidal anti-inflammatory drugs (NSAIDs), colchicine, conventional disease-modifying antirheumatic drugs (cDMARDs; methotrexate – MTX, cyclosporine A – CsA), biologic agents (adalimumab, anakinra, canakinumab and rituximab), immunosuppressants (cyclophosphamide, 6-mercaptopurine), interferon α (IFN-α), and radiotherapy [9, 52].
In the clinical approach to reported cases of HS, both NSAIDs and GCs have been employed, with the latter being administered systemically, orally, topically, and intra-articularly.
While systemic GCs have frequently been used either as mono- or combined therapy [8], their effects are generally temporary, with cutaneous lesions and inflammatory markers recurring upon dose reduction or discontinuation and ineffective on arthritis [27, 57]. The current recommended approach is to use GCs adjunctively with other immunosuppressants [54]. Topical GCs have proven ineffective in ameliorating hyperpigmentation and cutaneous thickening, contrary to their systemic counterparts [58]. Intra-articular GC injections have led to symptomatic improvement in affected joints, albeit with a localised effect [27, 51, 52].
On the other hand, the use of NSAIDs has yielded a partial improvement, contrasting with the subsequent development of bone deformities [46].
Among the cDMARDs, MTX has been the most extensively employed therapeutic option for managing cutaneous and rheumatological manifestations of HS, often in combination with other agents, notably tumour necrosis factor α (TNF-α) inhibitors (TNFi), CsA, and tocilizumab (TOCI; anti-interleukin-6). When used as a single medication, MTX has contributed to partial clinical regression of hyperpigmentation, joint stiffness, and arthritis in some cases [23, 49]. However, persistent arthritis may necessitate the addition of etanercept (TNFi) to the therapeutic regimen [49]. Methotrexate has also been administered in conjunction with adalimumab (TNFi), exhibiting significant improvement in joint and ocular manifestations [46].
Concomitant use of oral MTX and NSAIDs did not lead to a reduction in skin thickening [51]. Methotrexate has been explored with other agents. In a case series from the United States [27], one patient showed no clinical improvement after 4 months of subcutaneous MTX (12.5 mg/week), even after the addition of CsA (3–5 mg/kg/day). In contrast, signs of improvement in arthritis have been observed in another patient from the same case series after 6 months of treatment with subcutaneous MTX (25 mg/week). Furthermore, in the latter patient, the combination of oral MTX at the same dose with TOCI (162 mg) contributed to skin tone fading and normalisation of ESR.
Indeed, across multiple cases, TOCI consistently demonstrated remarkable efficacy in ameliorating skin manifestations. Thirteen out 16 patients treated with interleukin-6 (IL-6) antagonist experienced significant reduction in skin infiltration and thickening [12, 27, 39] and/or normalisation of inflammatory markers, particularly ESR and CRP [27, 39, 52]. Administered intravenously at a dosage of 8–12 mg/kg every 2 to 4 weeks, therapeutic effects have been evident as early as the first month of monotherapy [18], further consolidated at 2–3 months [23, 27], and sustained throughout a 14-month follow-up period [58]. Some patients have maintained this treatment for an extended period of 2 to 3 years, yielding consistent long-term benefits with disappearance of cutaneous lesions [23, 39].
These results primarily address the partial effect of IL-6 blockade on the overall clinical presentation of HS because treatment with TOCI has shown no definitive effect on articular lesions (hallux valgus, camptodactyly) even when co-administered with oral GCs [23]. However, Bloom et al. reported partial improvement in arthritis among their 5 patient cases following IL-6 antagonist infusions [27], and Lequain et al. [39] documented 2 cases of arthritis improvement with an absence of flares after 4 months of treatment. Intriguingly, mycophenolate mofetil at a dosage of 500 mg bis in die has demonstrated efficacy in resolving hyperpigmentation and skin induration, accompanied by a reduction in joint stiffness over an 18-month follow-up period [29].
A recent case investigated the combined treatment of a JAK inhibitor, baricitinib, and hydroxychloroquine (HCQ) in a genetically confirmed HS patient. Promising outcomes were observed after one year of treatment, including clinical improvement, reduced pain, enhanced functional capacity, and normalisation of inflammatory markers (CRP, ESR, and Inflammatory Score [IS]). Initially, the patient showed partial improvement with MTX (15 mg/week); however, the regimen was switched to HCQ combined with baricitinib, instead of TOCI. The use of HCQ is supported by its stabilising effect on immune cell lysosomes, dampening inflammation triggered by TLR receptors and IFNs due to adenosine accumulation in defective ENT3 cells. Meanwhile, the rationale for employing a JAK inhibitor lies in its broader action on both IFNs and IL-6, contributing to a slight alleviation of rheumatological manifestations [47].
In the context of identifying novel therapeutic strategies for HS, a potential candidate has emerged: the mitogen-activated protein kinase (MEK) inhibitor, a type of targeted therapy.
Enhanced MAPK signalling leads to inflammation and cytokine secretion, primarily attributed to mutated ENT3 monocytes. The MEK inhibitor exerts its therapeutic effect by reversing the upregulated expression of inflammatory cytokines in HS through inhibition of the TLR8-MEK-ERK signalling pathway. Consequently, MEK inhibitors like trametinib have demonstrated efficacy in resolving histiocytosis and inflammation, highlighting their potential as a promising therapeutic option for HS, even in cases resistant to other treatments such as anti-IL-6R antibodies [12].
Conclusions
H syndrome is a rare autoinflammatory syndrome with multisystemic manifestations. Osteoarticular involvement is frequently found in HS, but it is rarely reported in the rheumatological literature published to date.
This syndrome should be considered in patients with hallux valgus, short stature, fixed phalangeal contractures, arthritis, and systemic inflammation, mainly associated with cutaneous and endocrine manifestations.
Several treatments have shown to be effective in treating joint stiffness and arthritis, including MTX, TOCI, and JAK inhibitors. Mitogen-activated protein kinase inhibitors, introduced as a new therapy, have demonstrated very promising results. From a rheumatological perspective, it is crucial to note that once joint deformities have developed, these therapies become ineffective. Hence, early diagnosis is paramount to address the underlying inflammatory and immunological processes driving this syndrome and prevent the onset of permanent joint deformities. This case report is a very welcome contribution highlighting the importance of recognising HS among rheumatologists.