
Introduction
Residual pain (RP) is a major unmet medical need observed in patients suffering from rheumatoid arthritis (RA), which decreases their quality of life, even after achieving remission or low disease activity [1]. Lee et al. [2] reported that 47.3% of RA patients with low disease activity reported moderate to high levels of pain. However, in a different study Lee et al. [3] found that only 12.5% of patients who were at one year of remission had clinically significant pain (defined as Multi-Dimensional Health Assessment Questionnaire pain ≥ 4). McWilliams et al. [4] reported that one year after immunosuppression with disease-modifying antirheumatic drugs (DMARDs), the pain remained a problem for 40–50% of patients despite adequate suppression of inflammation. The data of RP prevalence among RA patients are inconsistent, yet the problem of RP among RA patients exists and is clinically significant. The aforementioned findings suggest that the gold standard treatment for RA, i.e. DMARDs, is not adequately suited to patients suffering from RP, and poses a question of whether RP should be a factor dictating the type of therapy chosen for patients. Furthermore, the fact that patients experience pain when in remission or low disease activity suggests that non-inflammatory mechanisms are involved in RP. Understanding these mechanisms is essential to effectively target therapy for affected patients.
This review aims to summarize the findings about types of pain, tools used to measure pain, and their accuracy, the factors that affect RP, mechanisms, comorbidities, and what methods of pain assessment could be helpful for patients with RP.
Pain mechanisms
Inflammatory pain
Inflammatory pain in RA arises from structural damage and peripheral sensitization induced by chemical mediators. Bone and cartilage damage is rapid and dynamic, and affects the majority of RA patients within the first year [5]. The continuous inflammatory attack of the synovial membrane on bone results in irreversible structural damage in RA such as joint space narrowing, periarticular osteoporosis and bone erosions [6]. Progressive destruction of cartilage and subchondral bone tissue causes chronic pain in RA. Peripheral sensitization is, according to the definition of the International Association for the Study of Pain [7], increased responsiveness and a reduced threshold of nociceptive neurons in the periphery to the stimulation of their receptive fields. It is induced by the interaction between the immune cells and the nociceptor (Aδ and C fibres are the two main types of primary afferent nociceptors) via inflammatory mediators including cytokines: tumour necrosis factor α (TNF-α), interleukin-1β (IL-1β), IL-6, IL-17, chemokines, nerve growth factor and prostaglandins [8]. They are produced by resident and infiltrating immune cells during joint inflammation, but also arise from cell destruction (hydrogen ions) or are delivered by the circulation (bradykinin) [9]. These inflammatory mediators bind to specific receptors on the nociceptors, which results in activation of intracellular signalling pathways, leading to a phosphorylation cascade, as well as to increased expression of sodium and transient receptor potential channels [10]. This process causes hyperexcitability of the sodium channel and makes other receptors, such as transient receptor potential vanilloid-1 (TRPV1) and transient receptor potential ankyrin-1 (TRPA1), hypersensitive [11]. This is shown in Figure 1. Among the infiltrating immune cells are mast cells, which in an inflammatory environment release on degranulation histamine and serotonin [12]. Histamine activates H1 and H2 receptors expressed on nociceptors, which results in increased expression of Nav1.8 channels and hypersensitivity to noxious stimuli [13]. The inflammatory mediators, along with tissue acidification, act synergistically to increase the activity of nociceptive primary afferent neurons and reduce their threshold to generate action potentials. Peripheral sensitization clinically manifests as peripheral hyperalgesia [14].
Fig. 1
Pain in inflammatory and non-inflammatory state in RA. In the inflammatory state resident and infiltrating immune cells release inflammatory mediators that act on their respective receptors reducing the response threshold of Nav and TRP channels, and activating nociceptive primary afferent neurons. In the non-inflammatory state nociceptive signals in the dorsal horn of the spinal cord lead to release of glutamate, substance P, and calcitonin gene-related peptide, which bind to their respective receptors on the second order neurons, and lead to heightened excitability of the neurons; central sensitization. Figure based on information provided in book by Biddle et al. [91] and article by Buch et al. [92].
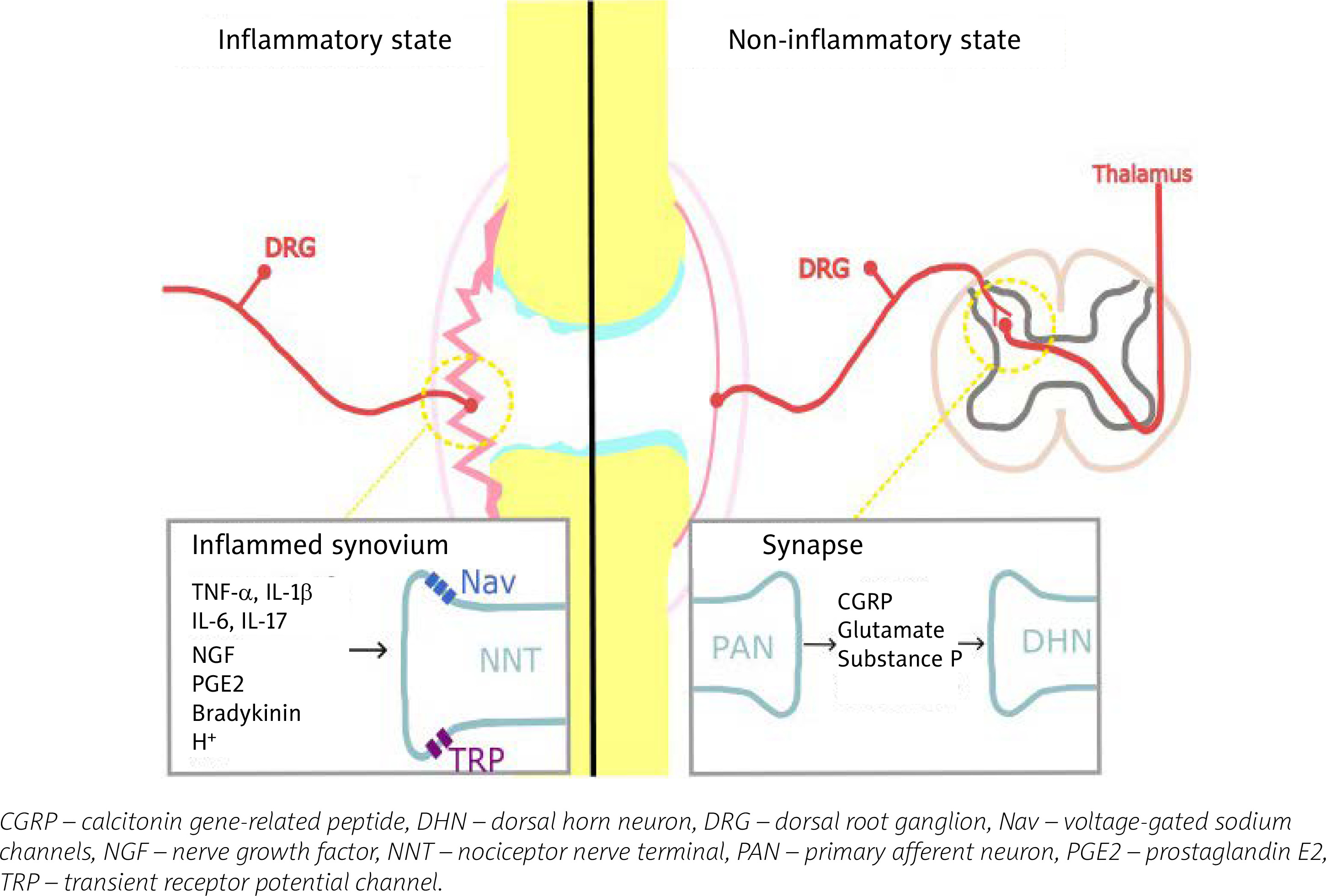
Neuroinflammation is currently being investigated as a mechanism of pain development in RA. Neuroinflammation is defined as inflammatory mediator release induced by glial cell activation [15]. The propagation of peripheral inflammatory mediators across the blood-brain barrier to the central nervous system (CNS) occurs through the interaction of the peripheral immune system with the CNS myeloid cells, which include microglia and perivascular macrophages [16]. Spinal astrocytes and CNS myeloid cells are activated and release pro-inflammatory mediators such as IL-6 and IL-1β, which are known to directly sensitize nociceptors and mediate pain [17–19]. In the brain activation of glial cells, such as astrocytes, in different brain regions (pre-frontal cortex, the primary somatosensory cortex or anterior cingulate cortex) leads to dysregulation of glutamate and γ-aminobutyric acid (GABA), causing an imbalance of excitatory and inhibitory neuronal inputs, which enhances pain signals [18, 20, 21].
Non-inflammatory pain
Research by McWilliams et al. [22] revealed a discrepancy between the assessment of inflammation and patients’ reported pain, with 64% of participants in pain flare not concurrently in Disease Activity Score with 28-joint count (DAS28) flare and 60% of those in DAS28 flare not concurrently in pain flare. Patients with pain caused by other than inflammatory mechanisms can be overtreated with DMARDs, which in the case of these patients is not only ineffective but can also unnecessarily expose them to the risk of adverse events. Clinically, pain secondary to an inflammatory flare must be differentiated from pain secondary to central sensitization as they require vastly different management approaches [10].
One of the postulated mechanisms of non-inflammatory pathogenesis of pain is central sensitization. Central sensitization is, by definition of the International Association for the Study of Pain [7], increased responsiveness of nociceptive neurons in the CNS to their normal or subthreshold afferent input. Continuous nociceptive signals in the dorsal horn of the spinal cord lead to the release of glutamate, substance P and calcitonin gene-related peptide. Glutamate binds to its (N-methyl-D-aspartate (NMDA) receptor and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor/kainate) receptors on the second order neurons. This binding results in an influx of intracellular calcium, activation of various kinases, and translational, post-translational, and transcriptional changes resulting in changes in gene expression [23]. Other mechanisms involved in central sensitization are microglial activation and disinhibition. Under normal circumstances, inhibitory interneurons modulate pain transmission by releasing GABA and/or glycine in order to decrease the excitability of first order neurons. However, when there is peripheral injury, this inhibition can be lost, resulting in hyperalgesia. The continuous release of adenosine 5′-triphosphate (ATP) and several other chemokines in the dorsal horn of the spinal cord can activate microglial cells, which, in turn, release brain-derived neurotrophic factor. The factor promotes hyperexcitability and hypersensitivity in response to both noxious and innocuous stimulation, i.e. hyperalgesia and allodynia [24]. All of these processes collectively contribute to the heightened excitability of second-order neurons, referred to as central sensitization. Additionally, interactions between microglial cells, astrocytes and neurons in the dorsal horn of the spinal cord drive the production of pro-inflammatory cytokines and chemokines that increase second-order neuron hypersensitivity responsible for central sensitization [11].
Interestingly enough, cytokine inhibition, i.e. IL-6 inhibition in RA, has been associated with a reduction in non-inflammatory pain. In an analysis of clinical trial data, conducted by Choy et al. [25], non-inflammatory pain was common at the beginning of the study, and the percentage of patients with non-inflammatory pain after treatment was lower after sarilumab (which targets the IL-6 receptor) than with adalimumab (a TNF inhibitor) treatment. Additionally, a study by Ahmed et al. [26] found that 12-month treatment with abatacept (a CTLA-4 analogue) compared to adalimumab resulted in greater improvement in pain sensitisation. The superiority of other treatments compared to treatment with adalimumab regarding pain sensitization suggests that there is an as yet unrecognized systemic neuro-inflammatory component of pain in RA.
Yet another study found that the number of patients in remission or low disease activity with minimal or no pain was greater with baricitinib (Janus kinase inhibitor, JAKi) than placebo. Baricitinib, at a dose of 4 mg once daily, provided enhanced improvement in pain in patients with well-controlled RA, suggesting that it may produce effects beyond immunomodulation [27]. Furthermore, a meta-analysis by Dougados et al. [28] reported that patients with RA, who achieved abrogated inflammation with tofacitinib (JAKi) or adalimumab after 3 months had greater RP reduction than those receiving a placebo. This may imply that tofacitinib and adalimumab have analgesic benefits beyond those related to inflammation reduction. Clinical trials comparing JAK inhibitors and TNF inhibitors suggest that JAK inhibitors are associated with significant clinical improvements including pain [29, 30]. The promising effect of JAK inhibition may suggest that JAK-STAT signalling has a role in the pathogenesis of pain in RA.
Moreover, there are factors outside of inflammatory mechanisms, such as pain perception and initiation, that could lead to the development of residual pain. According to Bécède et al. [31], female sex is an important predictor of the refractory course of RA, which could possibly be related to the differences in how pain is perceived by men and women [32]. The same study found that delay of treatment initiation is a modifiable risk factor of refractory disease, while other studies documented poorer outcomes, greater disease progression, and a worse response to treatment in individuals who have delayed initiation of therapy [33–35].
Types of pain in rheumatoid arthritis
The pain in RA can be divided into acute (less common) and chronic pain. Acute pain usually appears during flare-ups and aggravation of disease activity, and commonly is considered to indicate a need for initiation, change, or increase in therapy [36]. Disease flares and acute pain can occur following a period of low disease activity or on the background of ongoing active disease and chronic pain. According to several studies, flares of inflammatory disease activity and pain often do not coincide [22, 37, 38]. Misinterpretation of RA flares of pain as a sign of uncontrolled inflammatory disease may result in patients being subjected to unnecessary or ineffective interventions, and could also impede patients from receiving more effective management options. The origin of pain flares can come from structural damage to the joint, overstrain of already damaged and affected joints, or other reasons completely (see below for comorbidities).
In a chronic inflammatory process resistant to DMARDs (difficult to treat disease – DTT) or without proper anti-inflammatory treatment, the neuropathic component may be involved. Koop et al. [39] reported that despite low disease activity, according to DAS28, 44% of patients still reported clinically significant pain. Out of them, 17% had likely neuropathic pain (NP), according to painDETECT, and 21% had possible NP features. Moreover, Ahmed et al. [40] obtained similar results, with 28% of patients having possible NP and 5% having features of likely NP measured using painDETECT scoring. The study by Perrot et al. [41] reported a relatively high percentage of patients (36%) with NP. This might be attributed to their use of the DN4 questionnaire, which is known for its high sensitivity but lower specificity in diagnosing NP [42].
A relatively new phenomenon described is nociplastic pain. Nociplastic pain is, according to the definition of the International Association for the Study of Pain [7], pain that arises from altered nociception despite no clear evidence of actual or threatened tissue damage causing the activation of peripheral nociceptors or evidence for disease or a lesion of the somatosensory system causing the pain. The pain arises from a change in function of nociceptive pathways. This change is linked to different mechanisms including central sensitization [43]. The nociplastic component of pain is present in both widespread conditions such as fibromyalgia syndrome (FMS), and localized conditions such as chronic temporomandibular pain disorders (TMDs), chronic primary bladder pain syndrome, irritable bowel syndrome (IBS), tension headaches, and chronic migraine headaches. Besides pain, these conditions often coincide with symptoms such as fatigue, memory issues, compromised sleep quality, and disturbances in mood [44].
Diagnosis and measures of pain in rheumatoid arthritis
Unfortunately, managing pain in patients with RA is a challenge, because pain can often be discordant or undetected with standard RA specific surveillance strategies, which focus on indicators of inflammation, such as examination findings, serological markers, and imaging [45, 46]. According to the 2019 update of the American College of Rheumatology (ACR), preferred clinical disease activity measures in RA include [47]:
Disease Activity Score with 28-joint count (DAS28),
Clinical Disease Activity Index (CDAI),
Simplified Disease Activity Index (SDAI).
The EULAR (European Alliance of Associations for Rheumatology) response criteria, which are used to define the degree of clinical disease activity as well as the response to treatment, classify individual patients as non, moderate, or good responders, depending on two factors: current DAS28 in the patient and improvement vs. baseline DAS28 [48]. To conclude, both European and American associations agreed that DAS28 is the gold standard for measuring disease activity. The scale takes into consideration joint tenderness, subjective assessment of disease activity, and elevation of acute phase reactants – all of which can be related to pain. Clinical remission or near remission is defined in the above measure via cut offs. However, even remission does not mean patients do not experience pain. For example, a study by Lee et al. [3] found that of the 157 patients who met the DAS28 remission criteria, 12.5% had pain scores above or equal to 4, which is considered clinically significant. That is why a separate assessment of pain is also important and may be useful in the case of changes in parameters indicating inflammation, with a simultaneous high value of the patient’s activity assessment on the VAS scale. Routine Assessment of Patient Index Data 3 (RAPID3) and Patient Activity Scale-II (PAS-II) are both multidimensional scales, and may be useful tools in pain assessment to use additionally to the recommended DAS28. The RAPID3 and PAS-II ask patients explicitly to rate their pain as part of the overall disease measure. To conclude, remission in the context of the DAS28 should be considered mainly as a measure of inflammatory disease activity, while pain should be categorized as a separate symptom to inflammation and should be dealt with separately using a scale appropriate for the patient.
Comorbidities influence of pain in rheumatoid arthritis
Rheumatoid arthritis patients can also suffer from other secondary pain syndromes that affect the musculoskeletal system, such as osteoarthritis (OA) and fibromyalgia. Fibromyalgia is associated with widespread musculoskeletal pain and symptoms such as fatigue, sleep fragmentation, depressed mood, and anxiety [49]. Pathophysiology of FMS is theorized to be connected to the presence of central sensitization to pain and deficits in endogenous pain inhibitory mechanisms [50, 51]. The result of these mechanisms can be seen in patients, who in comparison to healthy individuals have low thresholds and tolerance of pain, hyperalgesia and allodynia [52, 53]. Additionally, as mentioned above, patients with FMS also have concomitance of nociplastic pain. According to the EULAR and ACR (both institutions use the same criteria), in order to diagnose FMS in a patient the following conditions have to be fulfilled:
The widespread pain index (WPI) is 7, and the symptom severity (SS) scale score is 5, or WPI equals 3 to 6, and the SS scale score is 9.
Symptomatology has been present at a similar level for at least 3 months.
The patient does not demonstrate any other disorder that would otherwise explain the pain [54].
From 4.9 to 52.4% of patients with RA have concomitant FMS, which surpasses the 1–5% prevalence of FMS in the general population [55, 56]. The wide range in prevalence estimates suggest that in many patients, the diagnosis of fibromyalgia is not made. It is possible that if patients with FMS were diagnosed early and treated as recommended by EULAR then RP in RA would not be such a prevalent problem [57]. Patients with concomitant FMS rarely achieve remission or a low disease activity score [58, 59] and switch treatment more often, because of non-response to treatment [60]. This could lead to strategies of escalating the intensity of RA treatment. The aim of RA treatment is to control inflammation, and composite disease activity indices may be affected by non-inflammatory pain in RA patients with co-existing FMS. To avoid over-treatment with DMARDs, an assessment of FMS should be considered in RA patients who do not fulfil the remission criteria.
The combination of inflammatory joint diseases (RA) and non-systemic inflammatory joint diseases such as OA is not rare, especially with the older RA population [61]. Pain in weight-bearing joints suggests mechanical pain, and is a separate mechanism of RP that can be addressed separately from inflammation treatment, including through treatment of possible comorbid OA [62]. It was found that patients with RA and OA had a higher proportion of RP compared to patients with only RA (59% vs. 29%) even in the absence of clinical inflammation [63]. Additionally, the study proved an already existing theory that patients with late-onset RA might have a susceptibility to articular damage related to previous OA changes [64]. Interestingly, a review by Beswick et al. [65] indicated that 9% of patients after hip replacement and 20% after knee replacement report unfavourable long-term pain outcomes. This review indicates that the entirety of pain experienced in OA cannot be solely attributed to structural joint damage. Central sensitization has been suggested as an explanation for the pain, similar to RA, that is not attributable to joint damage [66].
Discussion
Clearly there is a non-inflammatory component to pain in RA. Several factors contribute to the onset and maintenance of pain in RA beside inflammation, i.e. neuropathic and nociplastic pain, and comorbidities. These factors result in residual, chronic pain in patients who are treated with DMARDs/biological drugs and seem to have immunologically well-controlled disease.
First of all, criteria used by EULAR and ACR to assess disease activity lack detailed measures of pain, which leads to misdiagnosing patients with residual pain. A separate assessment of pain may be especially useful for patients who do not experience negative changes in parameters indicating inflammation or even improvement, while simultaneously pointing to a high value on the VAS scale. Furthermore, these pain measures should help identify the mechanism of pain and distinguish nociplastic and neuropathic pain from inflammatory pain. Identification of the origin of pain would lead to treatments tailored specifically to individual RA patients, allowing the targeting of the pathogenetic pathways, which cause this pain, and help avoid over-treatment with DMARDs/biological drugs and often drug switching as well as severe adverse effects. Both treatments with synthetic (sDMARDs) and biologic DMARDs (bDMARDs), have adverse effects: sDMARDs quite often cause gastrointestinal distress (nausea, abdominal pain, diarrhoea), allergic reaction, bone marrow suppression, and hepatotoxicity [67], while bDMARDs may among other things cause susceptibility to infections and non-infectious pulmonary disease [68]. In Table I different methods of measuring pain and their benefits and limitations for RA patients with RP are presented.
Table I
Examples of benefits and limitations of methods to assess the pain and make a differential diagnosis
| Method of assessment | Description of the method | Benefits | Limitations |
|---|---|---|---|
| Doctor assessment [1] | Combination of medical history and physical examination: joints for swelling, redness and warmth, rash/nodules on skin and gait evaluation | ||
| Numeric Rating Scale (NRS) [71] | Pain screening tool used to measure pain intensity. Patient self-report scale, where a patient selects a whole number (0–10 integers) that best reflects their pain intensity, 0 representing “no pain” and 10 “the worst imaginable pain” |
| |
| Quantitative Sensory Testing (QST) [72, 73] | Assess somatosensation using a variety of stimuli (thermal and mechanical) and collecting data based on the subjective experience of these stimuli |
|
|
| Pressure Pain Threshold (PPT) [74–77] | Self-reported pain assessment scale that has 24 single-score questions ranging from 0 to 6 | ||
| Rheumatoid Arthritis Pain Scale (RAPS) [78, 79] | Self-reported pain assessment scale that has 24 single-score questions ranging from 0 to 6 | ||
| PainDETECT questionnaire [80–82] | A 9-item self-report screening questionnaire developed to detect neuropathic in chronic pain disorders |
| |
| Douleur Neuropathique en 4 Questions (DN4) [82, 83] | Combination of patient interview and patient examination with 10 questions. Distinguishes between neuropathic and nonneuropathic pain using seven items related to symptoms (burning, painful cold, electric shock, tingling, pins and needles, and itching), and three items related to the clinical examination (hypoesthesia to touch and prick and brushing). Scores ≥ 4/10 indicate neuropathic pain | ||
| Patient Activity Scale II (PAS-II) [84] | Composite index composed of VAS for pain, Patient Global Assessment of Disease Activity, and the abridged version of the Health Assessment Questionnaire (HAQ-II) |
| |
| Routine Assessment of Patient Index Data 3 (RAPID3) [85, 86] | Score based on an overall assessment of the disease by the patient, the level of pain, and the amount of physical disability. Each of the 3 individual measures is scored 0 to 10 |
| |
| Rheumatoid Arthritis Severity Scale (RASS) [87] | Designed for use by physicians on their own patients, consists of three visual analogue scales: disease activity, functional impairment and physical damage | ||
| Central sensitization inventory (CSI) [90] | Self-report screening instrument designed to identify patients who have symptoms that may be related to central sensitization or central sensitivity syndromes. The CSI consists of two parts. Part A includes 25 questions related to common CSS symptoms. Part B (which is not scored) determines whether the patient has been diagnosed before with certain CSS disorders or related disorders. A score of more than 40 indicates the presence of central sensitisation. |
|
|
Secondly, every patient with RP should be carefully assessed for the presence of coexisting FMS. A tool that is not part of criteria for diagnosing FMS, but could be used in clinical practice, is a self-administered questionnaire, the Fibromyalgia Rapid Screening Tool (FiRST), which according to Perrot et al. [69] has a sensitivity of 90.5% and a specificity of 85.7%. An alternative tool is the Fibromyalgia Survey Questionnaire, which is also a patient-based questionnaire. The additional diagnosis is crucial to improving the patient’s quality of life, because treatment for FMS differs from RA, and patients affected by FMS might have issues other than musculoskeletal ones that might affect the perception of pain.
Thirdly, the approach of medical professionals should shift from “treating” pain arising from RA to improving the patient’s quality of life. Nowadays we have to face more and more chronic “untreatable” diseases, which used to be acute and quickly lead to disabilities, or even death, but now are better controlled, yet the processes behind them are still not completely understandable. One of these diseases is RA. The evidence for the best option to treat the pain per se in RA is still inconclusive, and increasing the dosage of DMARDs may not always be a solution to remove residual or chronic pain in RA. Greater emphasis should be put on diagnosing the source and type of pain in RA and managing the pain in RA with concomitant drugs such as neuromodulation techniques, antidepressants, psychological treatment methods (e.g. mindfulness training, cognitive behaviour therapy), painkillers (with caution), physical therapy and, if necessary, using surgical procedures as a way to treat pain.
Conclusions
Overall, RP continues to be a substantial challenge for numerous individuals suffering from RA, contributing to psychological distress, fatigue, and a diminished quality of life. In RA pain results from a combination of joint inflammation, structural joint changes, neuropathy, pain centralization, and comorbidities, i.e. OA and fibromyalgia. Not all the mechanisms have been thoroughly explored yet, and more research is required. Furthermore, RP can develop a neuropathic and nociplastic component. It is crucial to accurately identify distinct pain types to tailor specific treatment for every patient. In order to identify the origin of pain, medical professionals should use a separate assessment of pain, additionally to the recommended disease activity measure criteria. Employing multimodal pain management strategies is essential to enhance patient wellbeing and functionality.