
Introduction
Recurrent fever always raises concerns among patients and their parents, leading to frequent medical consultations with different specialists and often posing a challenge for physicians. Various conditions, including malignancy, infectious diseases caused by viruses or other pathogens, autoimmune diseases, inborn errors of immunity (IEI), and autoinflammatory diseases, may be accompanied by periodic fever [1]. As a result, patients with recurrent fevers often undergo a lengthy diagnostic process to establish a diagnosis, and in some cases, the cause remains unidentified [2, 3].
Recurrent fever is defined by at least 3 episodes of unexplained fever within 6 months [1–3]. Important criteria for diagnosing recurrent fever syndromes include the duration of each fever episode, the interval between fever episodes, symptoms associated with each episode, such as rash, abdominal pain, arthritis or arthralgia, chest pain, and the child’s well-being between fever episodes [1, 3].
Systemic autoinflammatory diseases (SAIDs) caused by dysregulation of the innate immune system leading to systemic inflammation are one of the known causes of recurrent fevers [1, 4]. They can be monogenic, multigenic, or multifactorial. The most common monogenic autoinflammatory diseases include familial Mediterranean fever (FMF), mevalonate kinase deficiency, familial cold autoinflammatory syndromes (FCAS), Blau syndrome, and others. Multigenic autoinflammatory syndromes in childhood include periodic fever with aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome, systemic juvenile idiopathic arthritis (sJIA), Behçet’s disease, and chronic recurrent multifocal osteomyelitis [5, 6].
Additionally, certain genes, particularly NLRP3, can cause three different diseases with varying severity and clinical manifestations: Muckle-Wells syndrome, FCAS, and neonatal onset multisystem inflammatory disease or chronic infantile neurologic cutaneous and articular syndrome [7].
Diagnosis is based on clinical presentation and genetic testing. To date, more than 50 genes associated with autoinflammatory diseases have been identified [7]. Systemic autoinflammatory diseases can have an autosomal-dominant or autosomal-recessive inheritance pattern. Specific diseases, such as FMF, can be inherited both autosomal-dominantly, usually with the M694del variant, and autosomal-recessively. However, in about 30–60% of patients, genetic tests cannot confirm the diagnosis, but they still have clinical manifestations [2, 8].
In the presence of clinical symptoms, particularly recurrent fever, in the absence of a molecular diagnosis and criteria for PFAPA, a diagnosis of undifferentiated recurrent fever (SURF) is justified [2, 8].
Timely diagnosis of autoinflammatory diseases is crucial for appropriate treatment and prevention of complications. The kidneys are one of the organs most affected by autoinflammatory and autoimmune diseases. Delay in diagnosis can lead to complications such as amyloidosis and subsequently renal failure. Nonamyloid-related damage can also affect the kidneys due to inflammasome activation, such as IgA nephropathy and different types of glomerulonephritis [9].
The aim of our study was to analyze the role of molecular diagnosis in the occurrence of recurrent fever syndrome in children, as well as to analyze the results of our molecular diagnostic research in children with recurrent fever and their correlation with clinical manifestations.
Material and methods
A case study is presented as a basis for the discussion: a single-center study that involved 39 children aged 1–17 years with suspected IEI, who underwent genetic testing. Among them were 12 children with recurrent fever syndrome.
Suspicion of IEI based on warning signs of primary immunodeficiency (PID), developed by the Jeffrey Modell Foundation (JMF) Medical Advisory Board [10] or other clinical signs of IEI [11], was the basis for referral for genetic testing. Sequence analysis and deletion/duplication testing of the 407/429 genes (Invitae Primary Immunodeficiency Panel) were conducted in the Invitae laboratory, USA, in 24 children. Testing was performed from 2020 to 2022. Veritas’ Customized Exome Panel, based on whole-exome sequencing with analysis of the variants of primary immunodeficiencies panel that includes 575 genes, was used in 15 children from 2023 to 2024.
Children with malignancy, recurrent fevers caused by viral infections, typical autoimmune diseases, and typical PFAPA syndrome [3] were not referred for genetic testing and, accordingly, were not included in the study.
A thorough analysis of the correlation between molecular diagnosis and clinical symptoms was conducted in patients with recurrent fevers, and in identifying autoinflammatory gene variants in other patients suspected of IEI.
According to the classification criteria for autoinflammatory recurrent fevers [3], a confirmatory genotype was considered pathogenic or likely pathogenic variants: heterozygous in autosomal dominant diseases, homozygous or compound heterozygous in autosomal recessive diseases. Variants of uncertain significance (VUS) were considered non-confirmatory genotypes. For FMF, a confirmatory genotype was considered to be a trans-compound heterozygote for one pathogenic MEFV variant and one VUS, or biallelic VUS, or heterozygous for one pathogenic MEFV variant. Benign or likely benign variants were excluded.
The study adhered to the guidelines outlined in the 1975 Declaration of Helsinki (revised in 2000) and received approval from the Ethics Committee of I. Horbachevsky Ternopil National Medical University, Protocol No 60 of September 1, 2020. Informed consent was obtained from all patients and/or their parents.
Descriptive statistics were used, with data presented as the mean and standard deviation (SD).
We performed a search in the PubMed Medline database using the specified search terms: “autoinflammatory recurrent fever”, “recurrent fever”, “undifferentiated recurrent fever”, “PFAPA” and “molecular diagnosis”. We included relevant full-text articles in English that were published between January 2014 and July 2024.
Case description
Among the 39 children with suspected IEI who underwent genetic testing, there were 21 boys (53.8%). The age of the patients ranged from 1 to 17 years, with an average age of 8.6 ±5.2 years. The JMF warning signs of PID were observed in 15 (38.5%) children, while the rest had other signs of IEI.
Genetic testing revealed from 1 to 13 variants in the PID genes in each patient; most of these variants were VUS. Pathogenic or likely pathogenic variants were identified in 19 (48.7%) children. Ten of them were diagnosed with primary immunodeficiency, while the rest were carriers of pathogenic or likely pathogenic variants. In total, 19 children (48.7%) had variants in genes associated with autoinflammatory-related disorders, and only 2 children had pathogenic or likely pathogenic variants in IL36RN and MEFV genes, 3 children had an increased risk allele in NOD2, and the rest had VUS. The most frequently observed variants were in the MEFV gene (4 children) and NOD2 gene (3 children).
One 16-year-old patient with recurrent abdominal pain and recurrent edema was suspected of and diagnosed with hereditary angioneurotic edema (HAE). Detection of a pathogenic variant in the SERPING1 gene confirmed the HAE diagnosis. However, the boy was also found to have a heterozygous missense variant in the MEFV gene (c.2082G>A (p.Met694lle)), classified as pathogenic and associated with MEFV-related disorders, although he currently shows no signs of the disease.
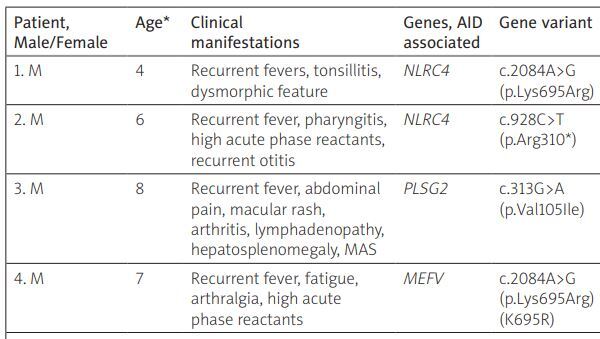
Recurrent fever was observed in 12 patients, of whom 7 were boys (58.3%). The age of the patients ranged from 1 to 16 years, with an average age of 8.5 ±3.9 years. The analysis of the identified genes associated with autoinflammatory-related disorders in patients with recurrent fever and their correlation with clinical symptoms is presented in Table I.
Table I
Clinical signs and detected genes associated with autoinflammatory-related disorders in patients with recurrent fevers
| Patient, Male/Female | Age* | Clinical manifestations | Genes, AID associated | Gene variant | Variant | Zygosity | Inheritance pattern | Disease associated with gene | Patient’s diagnosis |
|---|---|---|---|---|---|---|---|---|---|
| 1. M | 4 | Recurrent fevers, tonsillitis, dysmorphic feature | NLRC4 | c.2084A>G(p.Lys695Arg) | VUS | Het | AD | FCAS, MAS | PFAPA |
| 2. M | 6 | Recurrent fever, pharyngitis, high acute phase reactants, recurrent otitis | NLRC4 | c.928C>T(p.Arg310*) | VUS | Het | AD | FCAS, MAS | PFAPA |
| 3. M | 8 | Recurrent fever, abdominal pain, macular rash, arthritis, lymphadenopathy, hepatosplenomegaly, MAS | PLSG2 | c.313G>A (p.Val105Ile) | VUS | Het | AD | PLCG2-associated antibody deficiency and immune dysregulation (PLAID), FCAS, autoinflammation | sJIA, MAS, epilepsy |
| 4. M | 7 | Recurrent fever, fatigue, arthralgia, high acute phase reactants | MEFV | c.2084A>G(p.Lys695Arg)(K695R) | VUS | Het | AD, AR | FMF | SURF |
| 5. F | 11 | Recurrent fever, ILD, anemia, lymphopenia, hypogammaglobulinemia | SAMHD1 | c.8G>T(p.Arg3Leu) | VUS | Het | AR | AGS, FCL | Uncertain hypogamma-globulinemia, ILD |
| 6. F | 10 | Recurrent fever, stomatitis, gingivitis, cyclic neutropenia, constipation | SRP54^ | c.349_351del (p.Thr117del) | P | Het | AD | SRP54 deficiency | SRP54 deficiency, cyclic pattern |
| 7.F | 8 | Recurrent prolonged fever, rash, arthralgia | ND | – | – | – | – | – | sJIA |
| 8. F | 16 | Recurrent fever, recurrent pericarditis | ND | – | – | – | – | – | Recurrent pericarditis |
| 9. F | 15 | Recurrent fever, high IgE, PFAPA up to 7 year | ND | – | – | – | – | – | Post-COVID |
| 10. M | 8 | Recurrent fever, IBD, proteinuria | IFIH1 | c.2807+1G>A | VUS | Het | AD | AGS, Singleton-Merten syndrome | IBD |
| 11. M | 7 | JIA, resistant to treatment, recurrent fever, ADHD, hypertension, seizures, chronic adenoiditis, growth retardation, recurrent pneumonias | IL36RNSH3BP2 | c.338 C>T(p.Ser113Leu)c.671 A>G (p.His224Arg) | LPVUS | HetHet | ARAD | Pustular psoriasisCherubism | JIA, psychogenic fever |
| 12. M | 2 | Recurrent fever, rash, high inflammatory markers, SB, hydrocephalus | ND | – | – | – | – | – | uSAID |
AD – autosomal dominant, AGS – Aicardi-Goutieres syndrome, AID – autoinflammatory disease, AR – autosomal recessive, FCAS – familial cold autoinflammatory syndrome, FCL – familial chilblain lupus, FMF – familial Mediterranean fever, Het – heterozygous, IBD – inflammatory bowel disease, ILD – interstitial lung disease, LP – likely pathogenic, MAS – macrophage activation syndrome, NA – not available, ND – not detected, P – pathogenic, PFAPA – periodic fever with aphthous stomatitis, pharyngitis, and adenitis, SB – spina bifida, SURF – syndrome of undeferential recurrent fever, ADHD – attention deficit hyperactivity disorder, uSAID – undifferentiated systemic autoinflammatory disease, VUS – variant of uncertain significance.
No patient was found to have pathogenic variants in genes that could confirm an autoinflammatory disease. One girl (patient 6) with recurrent fever, stomatitis, gingivitis, and severe neutropenia in the blood test was found to have SRP54 deficiency with a cyclic pattern of neutropenia, causing recurrent fever every 2–3 weeks. This case was detailed in our previous publication [12].
Variants of uncertain significance in genes associated with autoinflammatory-related disorders with autosomal dominant inheritance were found in 6 patients. Two of them (patients 10 and 11) showed no correlation with clinical manifestations. In patients 1 and 2, VUS was identified in the NLRC4 gene with an autosomal dominant inheritance pattern. These patients had recurrent fever episodes resistant to antipyretic therapy, tonsillitis/pharyngitis, lymphadenopathy, and high acute-phase reactants during fever episodes. Additionally, patient 1 had dysmorphic facial features, and patient 2 had recurrent otitis, which led to genetic testing. Exclusion of other causes of fever and meeting PFAPA criteria allowed for this diagnosis in both cases.
Patient 3 presented systemic symptoms: fever, abdominal pain, erythematous rash, lymphadenopathy, arthralgia, hepatosplenomegaly, elevated acute-phase reactants, and no arthritis. Sudden hormone withdrawal and infection led to the development of macrophage activation syndrome (MAS) with coma. Additionally, the boy had neurological disorders with epilepsy, prompting genetic testing. A VUS in the PLSG2 gene, associated with FCAS and autoinflammation, could have influenced the disease’s development.
Patient 4 had 3 episodes of antipyretic-resistant fever accompanied by a hyperinflammatory response (high erythrocyte sedimentation rate [ESR] and C-reactive protein [CRP]) over 6 months. The first high fever episode occurred three weeks after pneumonia, possibly caused by COVID-19 (positive serology). Apart from high fever, resistant to non-steroidal anti-inflammatory drugs (NSAIDs), the boy had arthralgia, fatigue, and high acute-phase reactants. Multisystem inflammatory syndrome (MIS) associated with COVID-19 was ruled out. Subsequent febrile episodes occurred 2–3 weeks after mild viral infection with low-grade fever. The patient’s family history is unremarkable. Complete blood count showed moderate lymphopenia, neutrophilia, and elevated ESR and CRP during fever episodes. Between episodes, ESR and CRP were normal. Immunoglobulin levels and lymphocyte subpopulation markers were unremarkable except for elevated immunoglobulin (Ig) E (589 IU/ml). The boy is also being followed up for bronchial asthma and receives inhaled glucocorticosteroids (GCs) for control. Genetic examination revealed a missense variant in the MEFV gene (c.2084A>G), known as K695R and classified as VUS. According to the Eurofever/PRINTO classification criteria for recurrent fevers, particularly FMF, our patient lacked the confirming MEFV genotype and needed two of four signs: fever duration of 1–3 days, chest pain, abdominal pain, arthritis. Our patient only had arthralgia, precluding a FMF diagnosis at this stage. Thus, the boy was diagnosed with SURF.
Patient 5 had periodic fever with pronounced symptoms of interstitial lung disease (ILD), lymphopenia, and hypogammaglobulinemia. Immunosuppressive therapy with GCs and intravenous immunoglobulins had a positive initial effect on the course of the disease, but led to severe osteoporosis with vertebral compression fractures. Genetic testing did not detect any confirmatory genotype. The girl died of progressive ILD.
Patient 7 presented with a severe course of sJIA, and patient 8 presented with recurrent pericarditis. No gene variants associated with autoinflammatory diseases were detected in them.
Patient 9 had recurrent fever episodes predominantly in September and February. These fever episodes first appeared after COVID-19. Apart from fever, headache, and elevated CRP and ESR, no other inflammation signs were observed. Immunoglobulin E was consistently high (1,000–3,700 IU/ml) without significant allergy symptoms. Notably, COVID-19 IgM remained elevated for over a year. Up to 7 years old, the girl had classic PFAPA syndrome. Unexpectedly, no variants associated with autoinflammatory disorders were found in the girl.
Patient 12 with spina bifida and hydrocephalus was admitted with prolonged fever, rash on the trunk and limbs, high leukocytosis (up to 42 × 109/l), neutrophilia, thrombocytosis, elevated acute-phase reactants (CRP over 100 mg/l, ESR up to 54 mm/h). A high level of COVID-19 antibody titers was detected. Sepsis, shunt infection, urinary tract infection, MIS-C, autoimmune diseases, and allergic rash were ruled out. The first fever episode lasted over a month, poorly managed by NSAIDs, and recurred. Glucocorticosteroid therapy controlled fever episodes but not the rash, which periodically recurred. The child was referred for genetic testing for autoinflammatory disease. No autoinflammatory disease genes were found. The boy continues systemic GCs, with no fever episodes but periodic rash exacerbations. Acute-phase reactants show positive dynamics.
Discussion
The development of molecular diagnostics has not only increased the capabilities for diagnosing rare diseases but also posed more challenges for doctors, especially regarding the significance of VUS in diagnosing various diseases and the influence of these variants on the clinical manifestations of diseases. Every year, new genes associated with autoinflammatory diseases and other IEI are discovered [7].
For instance, FMF was previously considered a disease inherited in an autosomal-recessive pattern. However, today, there is substantial evidence for autosomal-dominant inheritance, particularly for the M694del variant [13]. The 2019 IEI classification notes that the MEFV inheritance pattern can be both autosomal dominant and autosomal recessive [14]. Even within one family, different clinical manifestations and varying gene expressivity can be observed [15, 16]. Regarding our four patients with identified variants in the MEFV gene, among whom three had VUS and one had a pathogenic variant, all were heterozygous. Only one patient exhibited symptoms of recurrent fever (patient 4). This variant has been observed in FMF patients and other autoinflammatory diseases in both homozygous and compound heterozygous states [15, 17, 18], and in a compound heterozygous state in asymptomatic individuals [18]. In most cases, the variant had a mild effect or reduced penetrance [13, 17]. Moreover, several individuals with the p.Lys695Arg variant in a heterozygous state without a second pathogenic variant exhibited clinical symptoms of FMF [19, 20]. This variant is listed in ClinVar and classified as pathogenic/likely pathogenic by 13 submitters, as a variant of uncertain significance by 9 submitters, and as a probable benign variant by one submitter. According to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology criteria, the p.Lys695Arg variant was classified as a variant of uncertain significance for MEFV-related disorders [21]. Our patient did not have sufficient clinical criteria for FMF diagnosis at this stage, but the gene could play a role in the development of hyperinflammation, and the SARS-CoV-2 infection might have triggered its development. A recent publication demonstrated that in vitro functional analysis of pyrin inflammasome activation can distinguish SURF from FMF and PFAPA patients, indicating the involvement of the pyrin inflammasome in SURF’s pathophysiology [22].
The genetic basis for the development of PFAPA syndrome is recognized by many researchers, considering familial cases of the disease. However, a thorough exome analysis did not reveal common genes associated with PFAPA [23]. The authors concluded that the PFAPA syndrome might have high genetic heterogeneity. This hypothesis is supported by other studies investigating the role of autoinflammatory genes, particularly AIM2, MEFV, NLRP3, and MVK, in the development of PFAPA [24]. Researchers found various MEFV and NLRP3 gene variants and concluded that PFAPA is a multifactorial disease with low-penetrance effects in different genes combined with epigenetic and environmental factors, leading to a homogeneous clinical picture. Another study examined MVK, TNFRSF1A, and MEFV genes in PFAPA patients [25]. Regarding the NLRC4 gene, whose VUS was found in our patients 1 and 2, its role in PFAPA development has not been studied. However, pathogenic variants of this gene cause NLRC4-MAS and FCAS4, which are defects affecting the inflammasome [7]. NLRC4 is an important regulator of innate immunity, detecting bacterial virulence factors. The NLRC4 inflammasome is activated by flagellin or without it during bacterial exposure, leading to increased IL-1β, IL-18 secretion, and macrophage activation [7, 26]. Literature data indicate the development of recurrent fever, rashes, and other symptoms in patients with various NLRC4 gene variants [26, 27]; hence it would be interesting to further investigate the role of this gene in PFAPA development.
In general, the characteristic clinical picture and good physician awareness make diagnosing PFAPA syndrome straightforward [28]. However, an atypical disease course requires genetic testing to exclude other autoinflammatory diseases.
Three patients were diagnosed with JIA, two of whom had the systemic variant, with one case complicated by MAS. Activation of the innate immune system plays a major role in the pathogenesis of sJIA, leading to cytokine release and systemic symptoms [6, 29]. Inflammasome activation also plays an important role in sJIA development [29]. Genomic studies have shown a wide range of genes associated with sJIA not found in other types of arthritis, confirming their different genetic backgrounds [30], although further research is needed to determine the role of specific genes in sJIA development. The role of the PLSG2 gene in sJIA development has not been studied yet, but considering its role in autoinflammation via IL-1 pathway activation [7], it can be assumed that this gene and its variant may play a role in fever and systemic manifestations development. A recent study [31] confirmed broader genotypic and phenotypic correlations of functional variants in PLCG2, including a novel form of inflammatory diseases in carriers of heterozygous variants, highlighting the need for further research into the role of these gene variants in inflammatory disease development. Today, sJIA is considered a multifactorial autoinflammatory disease.
Two patients with recurrent fever, who did not have a confirming molecular diagnosis and did not meet PFAPA criteria, were diagnosed with SURF (patient 4) or undifferentiated autoinflammatory disease (uSAID) (patient 12). These conditions are diagnoses of exclusion and require ruling out not only PFAPA but also other autoinflammatory diseases through molecular diagnostics [2]. Fever episodes in SURF recur less frequently, with most cases having a history of past infections, including streptococcal pharyngitis, otitis media, sinusitis, and/or pneumonia [1]. In our cases, it was COVID-19. Other authors note that NSAID therapy can be effective in most patients [1], but in our cases, patients required GC therapy – short-term in one case to manage fever episodes and long-term in the other, where the child is on prolonged GC therapy. Although SURF and uSAID are considered synonymous [32], in our first case, fever was the primary symptom without other significant signs, while in the second case, fever was accompanied by a rash, which is currently the dominant symptom. Therefore, in our opinion, the term uSAID is more appropriate in this case.
Each year, new data emerge on the contribution of novel variants of various genes to the development of autoinflammatory diseases [33]. Therefore, presenting new variants may be important for further research.
In 6 out of 11 (54.5%) of our children with recurrent fever (excluding the child with neutropenia), the triggering factor was COVID-19. Another child developed hyperinflammatory symptoms with interstitial lung disease after a viral infection episode a year before the COVID-19 pandemic.
Overall, COVID-19 and its consequences, particularly MIS-C, can mask autoinflammatory syndromes due to similar clinical symptoms: fever, rash, arthralgia or arthritis, pronounced acute-phase inflammatory markers [34–36]. On the other hand, viruses can trigger flare of autoinflammatory diseases [1]. Recurrent fever syndrome has been reported in patients with MEFV gene polymorphisms [37] after COVID-19. Inflammasomes activated in response to SARS-CoV-2 infection are also associated with COVID-19 severity. A clear mechanism by which the SARS-CoV-2 N protein promotes NLRP3 inflammation activation and induces excessive inflammatory reactions has been described [38]. Post-COVID or long-COVID symptoms also include hyperthermic syndrome, fatigue, weakness, arthralgia, myalgia [39], which can complicate the diagnosis of autoinflammatory diseases and lead to their overdiagnosis.
In general, diagnosing congenital recurrent fever syndromes can be challenging, especially during the first episodes when there is no specific periodicity of symptoms, or if there is no regularity. Increased awareness of congenital autoinflammatory diseases with recurrent fever syndrome will improve their diagnosis and allow for appropriate treatment, preventing complications such as renal involvement [40].
A limitation of this study is the small sample size, restricted to research within a single center. However, we believe that our results could be interesting and potentially expand the range of genes to study in PFAPA syndrome, JIA, particularly NLRC4 and PLSG2 gene variants. Accumulating data will allow further research to be planned.
Conclusions
Diagnosing diseases presenting with recurrent fever syndrome can be challenging, and genetic testing does not always confirm the diagnosis. In the presence of neutropenia, congenital neutropenia with a cyclic pattern should be excluded. The presence of various gene variants associated with autoinflammatory-related disorders, even with low penetrance, combined with epigenetic and environmental factors, may contribute to the clinical presentation of the disease. The SARS-CoV-2 infection can trigger recurrent inflammatory reactions. The prevalence of various gene variants, especially VUS, associated with autoinflammatory diseases is high (48.7%) among children suspected of IEI, but the causal role of each gene variant in disease development must be carefully confirmed.
Further research into the role of gene variants, including those with low penetrance, and other factors may help identify the causes of recurrent fevers and prescribe appropriate targeted therapy.