
Introduction
Rheumatic diseases encompass pathologies of the musculoskeletal system and connective tissue, of non-traumatic origin, affecting individuals of all ages, with a higher prevalence in women. While there is a wide variety of these diseases, they can generally be grouped into the following categories: arthropathies; systemic connective tissue diseases; dorsopathies; soft tissue disorders; osteopathies and chondropathies [1].
Juvenile dermatomyositis (JDM) is one of the rheumatic diseases affecting children, and its pathophysiology remains poorly understood [1, 2]. Initially defined as a microvasculopathy, its primary clinical manifestations include skin and muscle involvement, leading to rashes in exposed areas, pathognomonic skin lesions such as Gottron’s papules and heliotrope, symmetrical and proximal muscle weakness, calcinosis, metabolic alterations and other symptoms resulting from chronic inflammation [3, 4].
Current treatments for JDM primarily involve synthetic disease-modifying antirheumatic drugs (DMARDs), such as methotrexate (MTX), cyclosporine, and hydroxychloroquine (HCQ), often in combination with glucocorticosteroids (GCs). These immunosuppressants target various immune system mechanisms in a non-specific and systemic manner, leading to potential adverse events [5–8]. However, the most effective drug for addressing JDM’s clinical and laboratory manifestations remains nuclear.
Biological agents such as rituximab (RTX) and tumor necrosis factor inhibitor (TNFi) drugs have been explored as second-line treatments for specific clinical manifestations, though their efficacy in JDM is still uncertain [9].
This systematic review aims to clarify the therapeutic potential of immunobiological agents, specifically RTX and TNFi, compared to standard therapies in the management of JDM.
The primary objective is to evaluate and compare the efficacy of these agents in improving clinical outcomes. Additionally, the review seeks to assess the safety profiles of these treatments by examining the frequency and severity of adverse events, thus providing comprehensive analysis of their benefits and risks relative to conventional DMARDs and GCs.
Material and methods
The target population comprised children and adolescents, under 18 years old diagnosed with probable or definite dermatomyositis according to Bohan and Peter’s criteria [10]. Patients treated with emerging biological therapies (TNFi and anti-B cell agents) were compared to those receiving standard treatment. The doses and duration of treatments were extracted as described in the original studies. The decision to focus on RTX and TNFi was based on their prominence in current treatment approaches for JDM and the availability of substantial clinical data.
The control group included patients receiving standard treatment with DMARDs and GCs. Clinical improvement, the primary outcome, was measured using subjective assessments by patients and physicians, as well as the Visual Analogue Scale (VAS) for disease activity, when applied by the study authors for skin and muscle strength. Imaging tests detecting edema and inflammation in the affected muscle region and laboratory markers of activity were also considered. Additional measures included the Childhood Myositis Assessment Scale (CMAS), Disease Activity Score (DAS), Manual Muscle Testing (MMT), Myositis Disease Activity Assessment Tool (MDAAT) and Disease Activity Core Set Measure (CSM) scores.
Procedures
This review stems from previous research assessing on-label and off-label drugs for JDM treatment, forming a segment of that broader work.
The review included clinical trials – both randomized and non-randomized, cohort, cross-sectional and case-control studies – that enrolled subjects aged 0–18. Case reports, case series, reviews, editorials, conference abstracts and posters were excluded. No restrictions were placed on language or year of publication to ensure a comprehensive scope of research.
The databases searched included MEDLINE via PubMed, Latin American and Caribbean Health Sciences Literature (LILACS), Scientific Electronic Library Online (SciELO) and Cochrane Library. Medical Subject Headings (MeSH) were used to define the descriptors and Keywords. The search terms employed were “Juvenile Dermatomyositis”, “Therapy”, grouped by the Boolean operator “AND”, with an age filter (child, teenager, adolescent or 0–18 years).
The identified studies were registered on the Rayyan platform, an intelligent systematic review tool, to remove duplicates and facilitate the study selection process through consensus on inclusion and exclusion criteria. The selection process was conducted by two independent researchers who resolved disagreements through discussion [11].
Data analysis
The study was conducted between February 2023 to September 2024, when new studies were reviewed and included.
The quality of this study was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) criteria, which evaluate risk of bias, consistency of results, imprecision, indirectness and publication bias for each outcome of the included studies [12].
Cochrane tools for systematic review were employed to assess the bias in randomized studies (Rob) criteria and the Robins I tool (Risk Of Bias In Non-randomized Studies – of Interventions), of which the elements analyzed were: bias resulting from the randomization process, deviations from initial interventions, missing outcome data, outcome measurement and selection of reported results. Studies were classified as having low, high or uncertain risk of bias.
Descriptive analysis of the sample from the systematic review was conducted using means and standard deviations for continuous variables and medians, frequencies, interquartile ranges, and proportions for dichotomous variables, all presented with a 95% confidence interval.
Analyzed variables
This review collected both quantitative data (e.g. time of diagnosis, treatment duration, medication doses, treatment changes, inflammatory markers values, disease activity score values) and qualitative data (e.g. desired effects, clinical improvement, adverse effects, perceived quality of life) from published studies on JDM treatment.
Bioethical standards
The study was approved by the Research Ethics Committee of the University Center São Camilo and registered with PROSPERO (CRD42022353563), an international database of prospectively registered systematic reviews. The registration is available at: www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42022353563.
Results
Juvenile dermatomyositis is a rare disease, and clinicals trials involving children with JDM remain limited. In the following sections, we present the findings from studies on JDM treatment with immunobiological therapies.
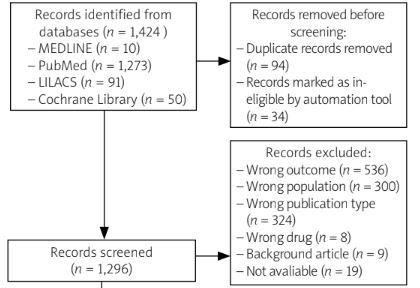
From the database search using the selected descriptors, 1,424 articles were initially identified. An automatic duplicate check using Rayyan software excluded 94 articles, followed by the removal of 34 articles for ineligibility. The researchers then carried out a title analysis and 1,296 articles remained for further screening.
Titles were reviewed independently by two researchers, who were blind to each other’s selection. This process led to the exclusion of 1,202 articles, with disagreements resolved through discussion. The main reasons for exclusion were: incorrect publication type (n = 324), wrong population (n = 300) and wrong outcome (n = 536). Additionally, 34 articles could not be retrieved, leaving 66 articles for abstract review.
Following the abstract review, 54 articles remained for full-text analysis, as 12 were excluded for not addressing the relevant outcomes or interventions required. Of these 54, 19 could not be retrieved, as they were not available in digital format. This left 35 articles that covered various treatments for JDM, ranging from conservative therapies such as prednisone to newer biologics. For this review, only studies involving RTX or anti-TNF therapies were included based on their prominence in current treatment approaches for JDM and the availability of substantial clinical data. After further screening, 30 articles were excluded, leaving 5 articles for detailed analysis.
The studies included were conducted in various countries, with two being experimental and 3 observational in design. Additionally, the selection process for the articles is illustrated in a flowchart following the PRISMA 2020 model, which can be seen in Figure 1.

Table I summarizes the clinical characteristics of the TNFi and B-cell therapies used in the included studies.
Table I
Overview of analyzed studies
| Author, year | Age | Previous medications | Treatment | Outcome measurement tools | Outcome | Robins | Grade |
|---|---|---|---|---|---|---|---|
| Oddis et al., 2013 [18] | 5 years of age or older | Glucocorticosteroids/azathioprine/methotrexate/mycophenolate mofetil/cyclosporine/tacrolimus/cyclophosphamide | Rituximab | MMT8, CSM | The majority of participants achieved clinical improvement in a similar way | Low | High |
| Aggarwal et al., 2016 [19] | Mean and SD: 15.3 (9.5) | Glucocorticosteroids/azathioprine/methotrexate/mycophenolate mofetil/tacrolimus/intravenous immunoglobulin | Rituximab | Myositis disease activity assessment tool (MDAAT), VAS, MDI and MDAS | Weaker tendency for faster improvement with early rituximab Global improvement in skin disease activity | Low | High |
| Bader-Meunier et al., 2011 [20] | First infusion of RTX: from 6.2 to 16 years | Glucocorticosteroids/plasma exchange therapy/immunosuppressive agents | Rituximab with glucocorticosteroids, immunosuppressors and plasmapheresis | Clinical improvement of the patients’ conditions | In 33% of the patients, clinical remission was complete. In 33% of patients, the course of the disease was unchanged | Critical | Moderate |
| Campanilho-Marques et al., 2020 [21] | Median age at disease onset: 5.2 (3.3–9.7) years | Glucocorticosteroids/azathioprine/methotrexate/cyclophosphamide/oral steroids/hydroxychloroquine | Infliximab or adalimumab | CMAS, MMT8, PGA and DAS | Infliximab improved global disease activity: PGA decreased (p = 0.005), skin involvement improved (p = 0.00043), modified DAS decreased at 6 months (p = 0.002), CMAS (p = 0.0002) and MMT 8 (p = 0.005) For patients who switched from infliximab to adalimumab (n = 15), there was an improvement in PGA | Moderate | High |
| Riley et al., 2008 [22] | Disease onset between 3 and 5 years | Glucocorticosteroids, azathioprine, methotrexate, cyclophosphamide, hydroxychloroquin, cyclosporin, intravenous immunoglobulins, pamidronate | Infliximab | VAS, CMAS and CHAQ | All patients showed great clinical improvement, with the greatest effect on muscle weakness, contractures and calcinosis | Low | Moderate |
[i] CHAQ – Childhood Health Assessment Questionnaire, CMAS – Childhood Myositis Assessment Scale, CSM – Disease Activity Core Set Measure, DAS – Disease Activity Score, MDAS – Modified Disease Activity Score, MDI – Myositis Damage Index, MMT8 – Manual Muscle Testing 8, PGA – Physician’s Global Assessment, VAS – Visual Analogue Scale
Initial treatment strategy
The initial approach to treating JDM usually involves GC, administered through various routes and doses. In Hasija et al.’s study [13], approximately 70% of the 275 patients showed clinical improvement and enhanced quality of life after 2 years of GC treatment. However, about 61% of patients were refractory to these drugs.
Similarly, Varnier et al. [14] reported that after 24 months of follow-up, 75% of the 127 patients had restored muscle strength and inactive disease. Studies by Sallum et al. [15] and Ng et al. [16] found that about 50% of cases showed a good response to GCs without needing additional medications. In Vaidehi et al.’s study [17], complete remission was observed in 42% of the 19 patients treated with GCs alone.
Despite the efficacy of GCs in the early stages of JDM, they are often insufficient for refractory or more complex cases. Only Varnier et al. [14] and Sallum et al. [15] reported not using additional medications in such cases. Therefore, in the search for safer and more effective treatments for refractory JDM, immunobiologics have emerged as a viable alternative.
Rituximab
The studies by Oddis et al. [18] and Aggarwal et al. [19] offer valuable insights into the effects of RTX on JDM, despite the small sample sizes. Notably, Aggarwal et al.’s publication [19] is an extension of Oddis et al.’s work [18], analyzing different outcomes with the same patient cohort. This makes these studies unsuitable for meta-analysis.
Both studies randomized participants into 2 groups: one receiving RTX early at diagnosis (0 to 1 week after initial screening) and the other receiving it later (8 to 9 weeks after screening). The study population included both adults and children with dermatomyositis, all of whom had disease refractory to traditional therapies.
Oddis et al.’ study [18] evaluated overall clinical improvement, using tools such as MMT, MDAAT and CSM. However, the results were not stratified by age group, making it difficult to draw age-specific conclusions.
The study [18] found no statistically significant difference between early and late RTX treatment in terms of clinical outcomes. Nevertheless, most participants in both groups experienced similar levels of clinical improvement. Infections were the most commonly reported side effect, though no difference was observed between the early and late administration of RTX.
Aggarwal et al.’s study [19] investigated the effects of the drugs on the cutaneous manifestations of the disease, using the MDAAT and the VAS for evaluation. The findings showed a slight trend toward faster improvement in skin symptoms, particularly rashes, among individuals receiving early RTX, although the effect did not reach statistical significance. Additionally, global skin disease activity improved in JDM patients, though there was little change in calcinosis. However, attributing these improvements solely to RTX is challenging, as patients were also receiving other medications during the study. Side effects, primarily infections, were consistent with those reported in Oddi et al.’s study [18].
These studies suggest that early treatment with RTX may slightly, although with weak evidence, improve the progression of the skin manifestations of the disease. It is important to note that no significant differences were observed in overall clinical outcomes between early and late interventions. However, the timing of treatment is not the only factor influencing disease progression. Other variables, such as individual patient characteristics and disease severity, play a crucial role in clinical improvement.
Despite the lack of significant differences in outcomes, delaying treatment is not recommended. Early intervention can reduce the severity of other disease manifestations and improve the patient’s quality of life. Further research with larger sample sizes is needed to better understand RTX’s role in JDM treatment.
Bader-Meunier et al.’s study [20] also evaluated RTX in a pediatric JDM cohort, focusing on its efficacy and safety. Muscle strength was measured using the MMT tool, while other manifestations, such as skin, joint and/or digestive involvement were assessed by physicians. Of the nine patients included, one discontinued treatment due to adverse events during the first RTX infusion.
Among the remaining 8 patients, 3 showed significant clinical improvement in both skin and muscle symptoms, while the others experienced an unchanged course of disease. Calcinosis did not improve with therapy and treatment regimens varied among participants, particularly with RTX and prednisone doses.
Regarding side effects, no infections were reported, although one case of intestinal perforation occurred, likely linked to methylprednisolone. However, this connection was questioned by the researchers. Most patients tolerated the therapy well, with only one experiencing a hypersensitivity reaction. Other adverse effects, such as hypogammaglobulinemia and neutropenia, commonly reported in the literature, were not observed, possibly due to underreporting or insufficient monitoring, as not all patients were actively investigated for such manifestations.
Tumor necrosis factor inhibitors
Both Campanilho-Marques et al. [21] and Riley et al. [22] investigated TNFi therapies in children with JDM who were refractory to traditional GC treatments, targeting a population with more severe disease.
Both studies used the CMAS to assess improvements in muscle symptoms. Campanilho-Marques et al. [21] also employed the MMT-8 and Physician’s Global Assessment (PGA) for patient assessment. To analyze the skin manifestations, they used the modified Skin Disease Activity Score (DAS). Riley et al. [22] used the Childhood Health Assessment Questionnaire (CHAQ) and VAS to measure clinical improvements, particularly in daily activities.
Campanilho-Marques et al.’ study [21] included patients treated with both infliximab (INF) and adalimumab (ADA), most of whom had previously received MTX, azathioprine or HCQ as monotherapy or in combination. Among the 39 patients who received only INF, significant improvement was observed in disease activity, as well as in muscle and skin manifestations. This was particularly true for 62% (24) of patients who had not previously used cyclophosphamide, as those with prior cyclophosphamide treatment tended to present more severe disease activity at the outset.
Regarding these patients, 38% (15) of the total switched from INF to ADA after the initial treatment, primarily due to treatment failure (especially skin manifestations) in the first few months or adverse effects such as hypersensitivity reactions. Despite this, these patients showed a significant improvement in disease activity, along with a improvement in the modified DAS scale and MMT-8 (p = 0.005) and in CMAS scale (p = 0.0002). In terms of adverse effects, mainly allergic reactions to INF, one case of sepsis and two of pneumonia were reported. Other adverse reactions were not severe, consisting mostly of mild infections.
As for Riley’s study [22], it included only patients who had previously undergone some form of treatment, mainly MTX and methylprednisolone. All 5 patients evaluated showed significant clinical improvement, with the greatest effect observed in muscle weakness, contractures and calcinosis. The GC dose was reduced in all cases and in 60% (3) of them, it was completely withdrawn. A side effect reported was an increase in lethargy and weakness before INF infusion, which improved when the time between sessions was reduced. Additionally, there was a reported case of calcinosis abscess infection.
The Childhood Arthritis and Rheumatology Research Alliance (CARRA) conducted a survey to reach a consensus on treatment decisions of JDM in different scenarios, predominantly in skin-related disease [23].
The 121 survey respondents were asked about the escalation pattern after 8, 12, and 16 weeks of treatment in moderate/severe JDM with skin manifestations that did not respond effectively. The vast majority preferred adding an agent to MTX rather than replacing it or making no changes.
Discussion
The findings from the analyzed studies suggest that MTX or GCs form the foundation of JDM treatment. For those opting for a change, mycophenolate was the most commonly selected non-biologic, particularly after 8 and 12 weeks. Interestingly, no other study identified mycophenolate as a major medication for treating the condition. However, the fact that all respondents were from a single region of the globe and a single institution might have introduced a regional bias. Most respondents favored adding an immunobiologic if initial therapy with one or two non-biologic DMARDs failed.
In another question, participants were asked to rank their preference among five immunobiologics after treatment failure, with RTX being the most frequently chosen, followed by abatacept (ABA), tocilizumab (TOC), and INF. Thus, RTX emerged as the most preferred treatment, supporting the findings of other studies.
Using a similar methodology, Tarvin’s research [24] aimed to explore the consensus on biologic treatment for patients with refractory moderately/severe JDM who did not respond to initial treatment with GCs, MTX, and intravenous immunoglobulins (IVIG). Four biologic drugs were included for selection in this survey: ADA or INF, ABA, RTX, and TOC.
For 76 CARRA members, RTX was the most frequently chosen, followed by ABA, ADA or INF, which corroborates the previous survey. Almost unanimously, 73 of the 74 participants who answered the question (99%) agreed that the patient should continue with at least one conventional DMARD and that the child should remain on their GC dose while receiving HCQ, GCs and IVIG.
Treatment of calcinosis as a complication of juvenile dermatomyositis
One of the main complications of JDM is calcinosis, which leads to calcification of damaged tissue and can occur anywhere in the body [25]. The presentation of calcinosis varies between patients, from plaques to calcified nodules. Most of the time, these formations are painless, but they can cause local pain and can limit movement, and lead to joint contractures [26]. When the calcium deposits break through the skin barrier, they create a favorable environment for bacterial infections, primarily by Staphylococcus sp. and Streptococcus sp., making these sites prone to recurrent infections and ulcerations [25].
The study by Campanilho-Marques [21] produced a significant finding regarding the efficacy of immunobiologics in treating calcinosis. Of the 72% (28) of patients with calcinosis in the study, 54% (15) showed significant improvement, with reductions in the size and number of skin lesions after starting TNFi.
Similarly, Riley et al. [22], who analyzed 5 patients with calcinosis, noted that the lesions became less painful, less hardened, and, in 4 out of the 5 cases, less extensive with the use of INF.
On the other hand, Aggarwal et al. [19] did not observe positive results with RTX as the introduction of this medication did not lead to any notable changes in this complication. Bader-Meunier [20] reached similar conclusions, reporting only minor clinical improvement in 3 patients, and no improvement in calcinosis for those who had it at the beginning of the study. Therefore, RTX was found to be ineffective in altering the course of calcinosis.
Barut et al. [27] reviewed the medical charts of patients diagnosed with JDM according to the Bohan and Carter criteria. All patients in this study received GCs, either orally or intravenously. A total of 96% (48 patients) were treated with GCs in combination with MTX. Among those who did not respond to GCs and MTX or became GC-dependent, 48% (23 patients) were additionally treated with cyclosporine. Other immunosuppressive therapies included intravenous IVIG (9), cyclophosphamide (5) and INF (2).
Among patients with calcinosis (19), 74% (14 patients) received cyclophosphamide, while 89.4% (17 patients) were treated with alendronate. Most of these patients showed a marked reduction in calcinosis, with all but two achieving complete remission. This suggest that cyclophosphamide and alendronate may be viable alternatives to biologics for treating calcinosis.
Compared to other therapies, immunobiologics appear to be a promising alternative to established treatments. In Sansone and Dubovitz’s study [26], which involved IVIG, some efficacy was observed, though the treatment’s effects were unpredictable and inconsistent, with side effects such as fever, nausea, and headache. In contrast, studies involving immunobiologics did not show the same variability in adverse effects, indicating greater stability in their safety profiles.
Although GCs remain the most standardized treatment for JDM, especially for milder cases, as highlighted by Giancane et al.’s study [28], the combination of prednisone with MTX or cyclosporine has significantly increased the incidence of infections in JDM patients. This raises expectations for immunobiologics as alternative treatments, especially for advanced or refractory cases, as they may offer better disease control while maintaining a similar adverse event profile [29].
Additionally, unlike Seshadri et al.’s study [30], which found no difference in controlling skin and muscle disease between high-dose and low-dose GCs, immunobiologics represent a promising alternative. In cases where initial treatment with prednisone is ineffective, immunobiologics have proven effective, particularly in controlling refractory forms of the disease.
Regarding the safety of the treatments analyzed, the drugs under review appear to offer a potentially safer and more consistent approach to treating JDM compared to other therapies. Analyzing the use of IVIG, Lang et al.’s study [31] demonstrated that IVIG was effective mainly in controlling cutaneous manifestations, while Tayfur et al. [32], despite reaching a similar conclusion, required joint therapy with bisphosphonates. Sansome and Dubowitz [26] raised concerns about the unpredictability of IVIG results, while Manlhiot et al. [33], despite affirming the safety of IVIG treatment, highlighted the adverse effects observed during the initial sessions.
Based on this systematic review, it is evident that treatment for JDM and its complications remains a controversial and poorly established topic. Numerous observations have been made over the years, but no clear consensus has been established on the most appropriate therapy. Furthermore, clinical trials on newer medications used to treat JDM complications are scarcer. Immunobiological drugs show promise as a treatment option but require further research to fully assess their impact on disease progression.
Nevertheless, by consolidating the findings from the studies analyzed in this review and comparing the effects of RTX and anti-TNF drugs, we propose a treatment flowchart, shown in Figure 2, based on the most robust evidence obtained.
Fig. 2
Flowchart for the treatment of the different clinical conditions of JDM and the treatment for its complications.
GCs – glucocorticosteroids, HCQ – hydroxychloroquine, INF – infliximab, IVIG – intravenous immunoglobulin, JDM – juvenile dermatomyositis, MTX – methotrexate.

The optimal treatment for JDM and its potential complications remains uncertain, largely due to the highly individualized response to drug therapies. Despite the limitations of the current studies, it can be inferred that for severe cutaneous and muscular disease, or in refractory cases, immunoglobulin and biologic therapies are often preferred. These medications are typically reserved for situations where standard treatment, such as prednisone, have not been effective. However, it is important to highlight that these drugs show limited efficacy in treating calcinosis.
While biologics offer potential benefits, they carry an increased risk of infection, emphasizing the need for clear and judicious criteria before initiating such treatments. Given these risks, further studies, especially randomized clinical trials, are required to accurately assess the effectiveness of these therapies. Currently, most publications on this topic consist of retrospective cohorts, case reports or series, often with small patient samples.
Conclusions
This systematic review underscores the potential of biological therapies, particularly with RTX and TNFi, in treating JDM. While GCs remain the cornerstone of initial treatment, many patients require alternative therapies. Rituximab has shown modest benefits, particularly for cutaneous manifestations, but further investigation is needed regarding its role in calcinosis. The TNFi, on the other hand, have shown more consistent results, particularly in improving muscle symptoms and reducing calcinosis, though adverse effects are a concern.
Early intervention with biologic therapies may offer benefits in managing refractory JDM and improving patient outcomes. Continued research with larger cohorts is essential to better understand the role of these emerging therapies, balancing their efficacy and safety.