
Introduction
Laugier-Hunziker syndrome (LHS) is an uncommon idiopathic disease, characterized by mucocutaneous, hyperpigmented macular lesions [1]. Although the etiopathogenesis is not fully known, tyrosine is thought to be responsible for the pathogenesis of enzyme hyperactivity in melanin biosynthesis [2]. With the exception of one familial case, consanguinity and genetic transfer have not been reported in the literature [3]. Mucosal pigmented lesions are usually seen located in the oral mucosa, the lips, cheek mucosa, and the hard and soft palate and more rarely in the gingiva, tongue and the base of the mouth. Nail involvement is seen in 50–60% of LHS cases, in the form of hyperpigmentation without dystrophic changes [4]. Dermatoscopic assessment is useful to show e.g. skin, nail or lips hyperpigmentation. However, the oral and esophageal mucosa or skin biopsy is more accurate and usually confirms the changes as basal cell hypermelanosis and acanthosis. Apart from cosmetic problems, the course of LHS is benign.
Laugier-Hunziker syndrome diagnosis is made clinically and histopathologically after exclusion of other potential causes. In the differential diagnosis, firstly Peutz-Jeghers syndrome, Addison’s disease, malignant melanoma, physiological melanoplasia and drug-associated pigmentations must be considered [1]. Although there are approximately 200 cases of LHS in the literature, to the best of our knowledge, there are no cases of LHS together with rheumatoid arthritis (RA) [5]. In this paper, the clinical characteristics are presented of LHS in a patient with RA.
Case report
The case concerns a 66-year-old woman with diagnosis of RA established 10 years ago, according to 1987 American College of Rheumatology criteria. In physical examination the patient also had longitudinal melanonychia in the nails of the hands and feet (Figs. 1 and 2) and brown pigmentations on the lips (Fig. 3) and in the pretibial region of the skin. Such skin, mucosa and nail changes appeared before the start of RA treatment (approximately 12 years previously). The described patient was taking methotrexate in the maximum tolerated dose (10 mg/week) and sulfasalazine (2 g/day) as RA treatment. There was nothing remarkable in the patient’s history or the family history. In the laboratory examinations, the peripheral blood cell count routine biochemical parameters and urine test results were all within normal limits.
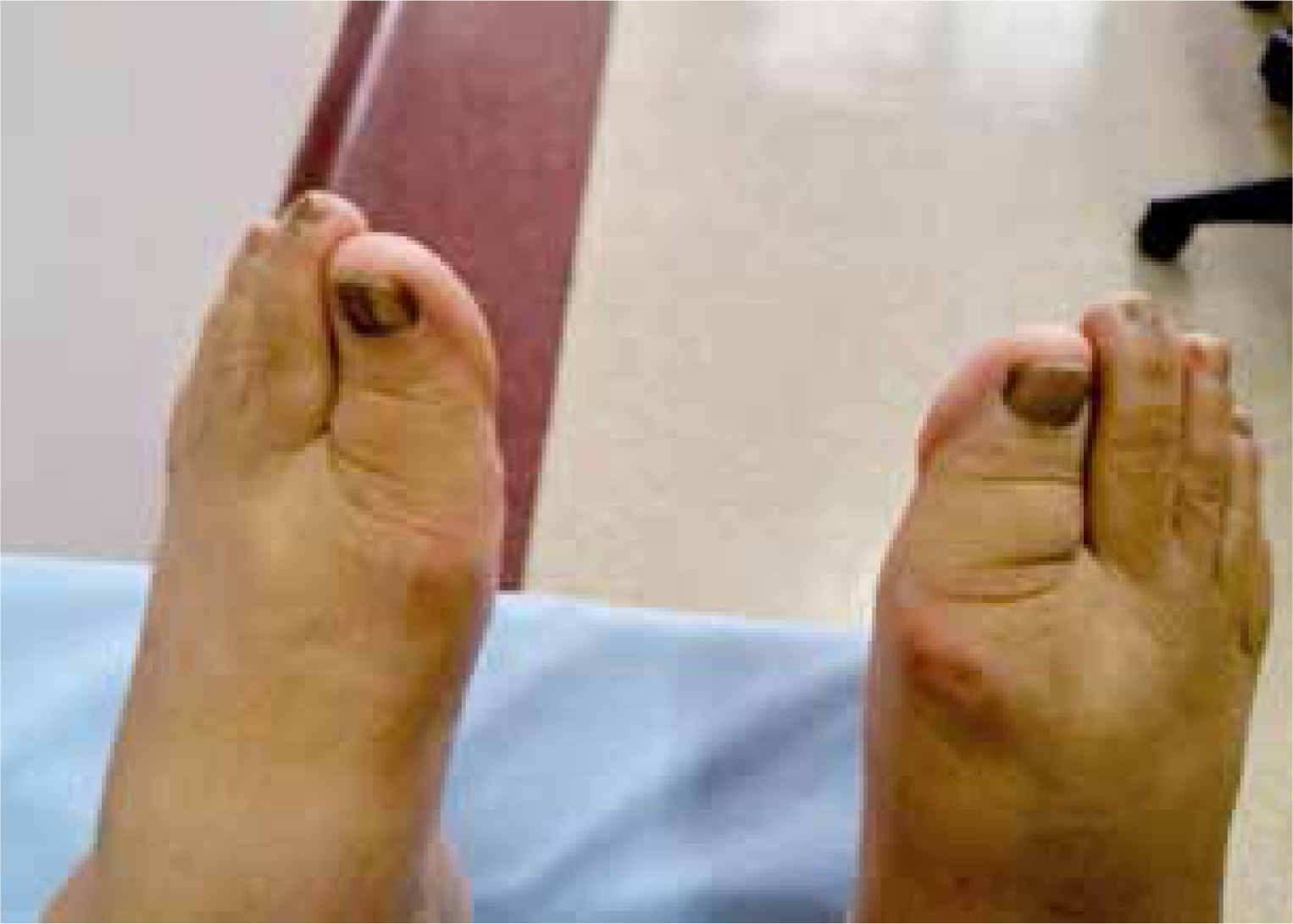
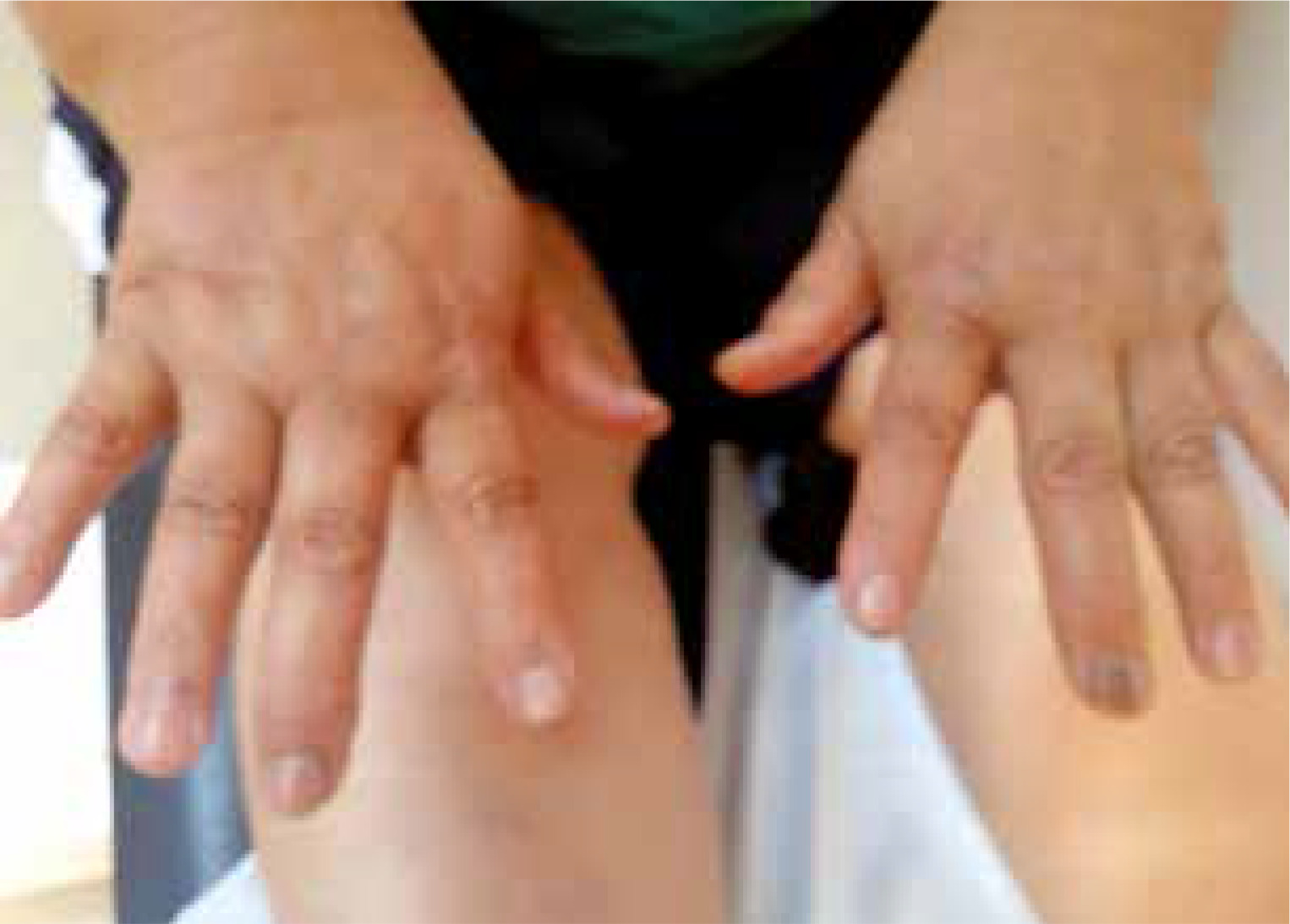
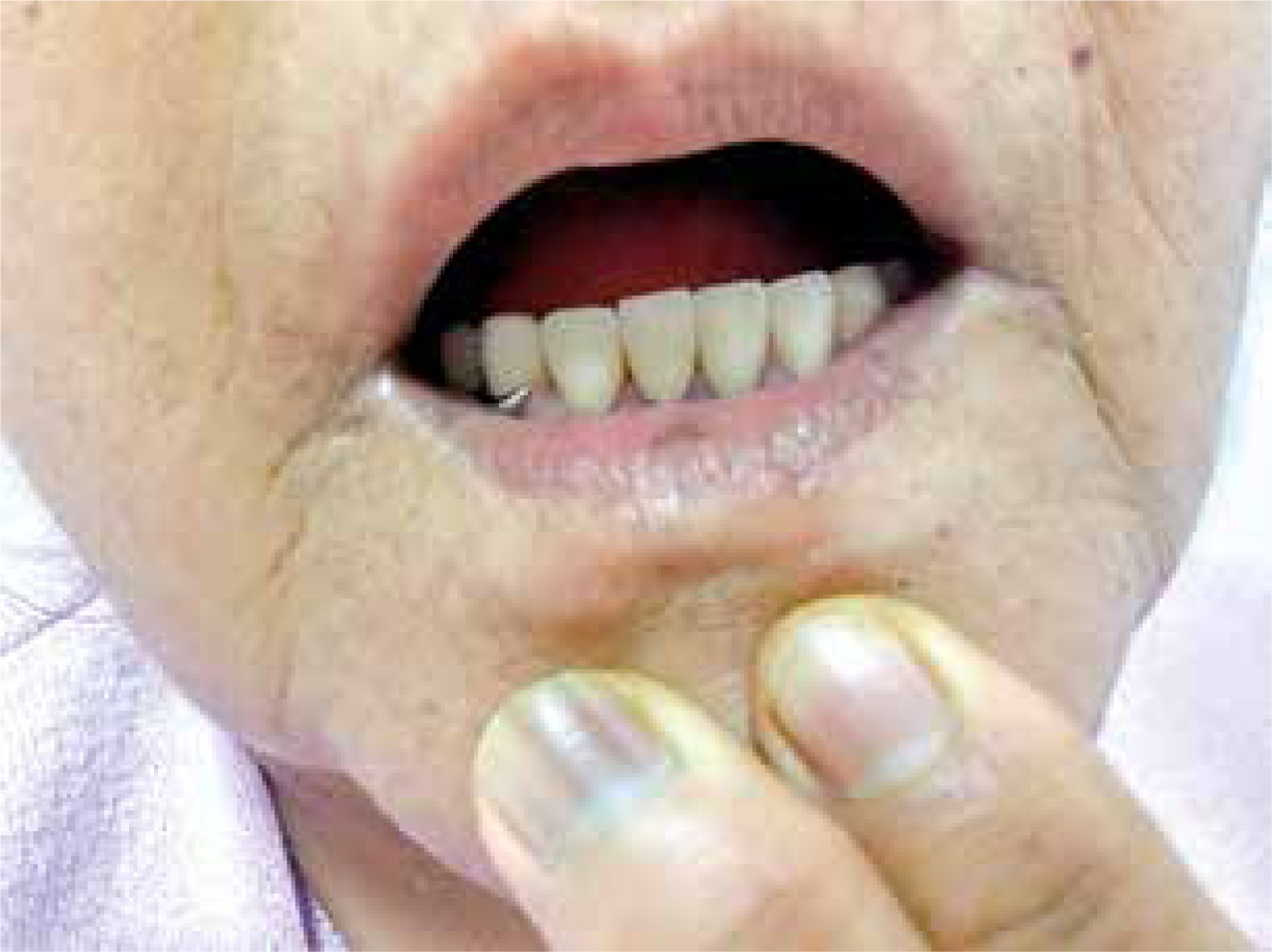
Rheumatoid factor, anti-CCP and antinuclear antibody profiles were negative, the erythrocyte sedimentation rate (ESR) was 21 mm/hour and the C-reactive protein (CRP) level was 0.9 mg/dl (normal range 0–0.5 mg/dl). Plasma cortisol levels, adrenocorticotropic hormone (ACTH) values and thyroid function tests (thyroid-stimulating hormone – TSH) were within normal limits. No pathological findings were determined in the comprehensive screenings applied in respect of malignancy and accompanying diseases, primarily abdominal ultrasonography, upper gastrointestinal track endoscopy, colonoscopy and mammography. As a result of a skin biopsy taken from the right pre-tibial region, increased melanin deposition was revealed in the basal layer (Fig. 4).
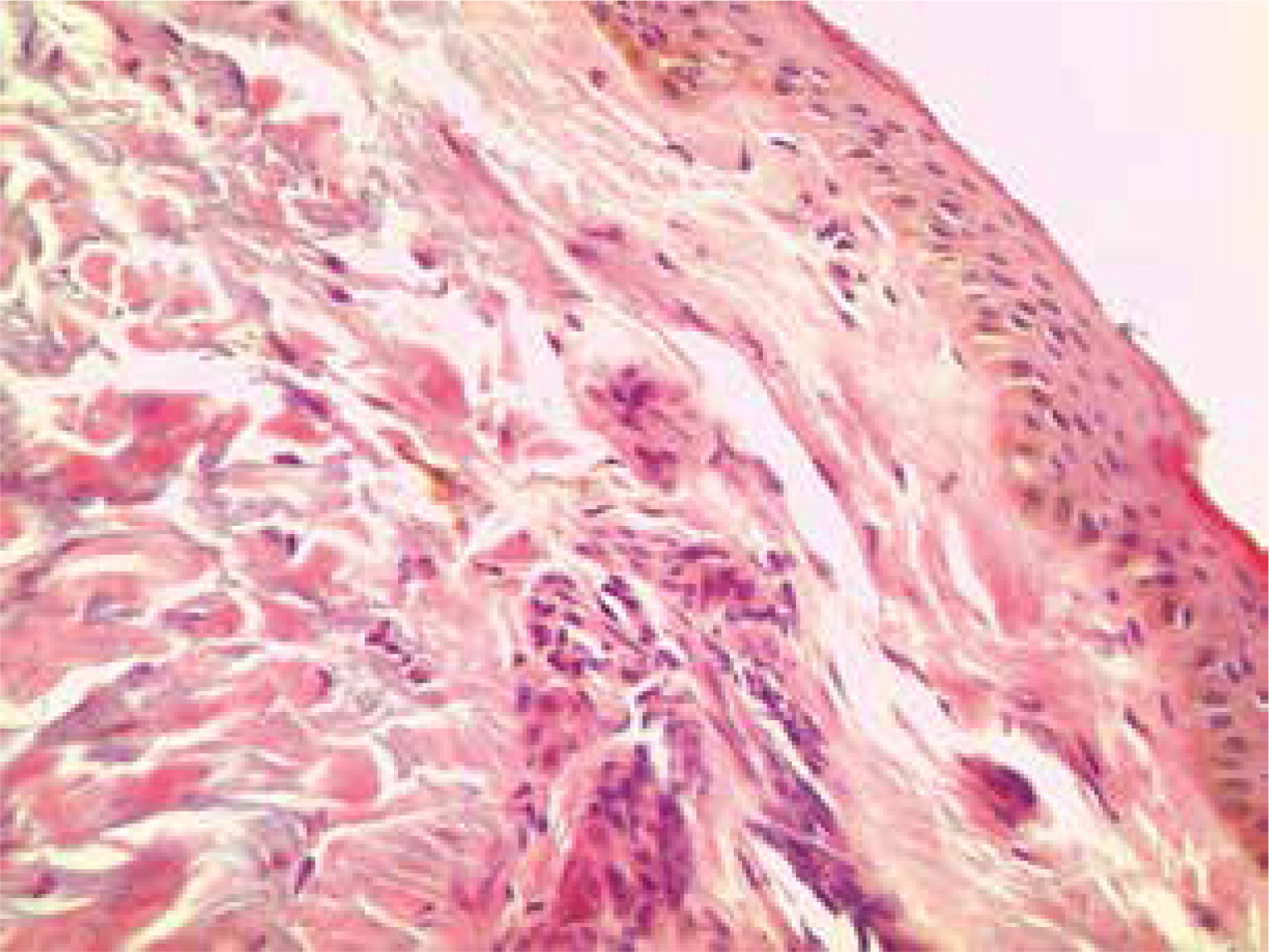
The diagnosis of LHS was made based on the clinical and histopathological findings of the evaluations made in the Dermatology Clinic.
Discussion
Laugier-Hunziker syndrome is an acquired disease, characterized by mucocutaneous hyperpigmentations, which was first described by Hunziker and Laugier in 1970 [6]. There are approximately 200 reported cases in the literature. The average age of the reported cases at diagnosis is 47.5 years old (range 12–87 years). Women are affected more frequently than men, with an overall female-to-male ratio of 1.8 : 1 [7]. The described case conforms to these epidemiological dependences.
Although the etiopathogenesis is not fully known, tyrosine is thought to be responsible for the pathogenesis of enzyme hyperactivity in melanin biosynthesis [2]. It has also been reported that there could be a relationship with autoimmunity and connective tissue diseases. In the literature, 2 patients with Sjögren syndrome have been determined with LHS but there are no reports of LHS together with RA [8, 9].
The diagnosis of LHS is made clinically and with the exclusion of other diagnoses. Focal, diffuse conditions and syndromes are included in the differential diagnosis of LHS [5]. Focal conditions are amalgam tattoo, graphite tattoo, hemangioma, melanoma, and nevus. Diffuse conditions are Addison’s disease, drug reactions and smoking. McCune-Albright syndrome, neurofibromatosis, Bandler syndrome, and Peutz-Jeghers syndrome (PJS) are considered in the differential diagnosis [5].
Malignant melanoma lesions, which should certainly be considered in the differential diagnosis, are larger, irregular and at risk of bleeding compared to benign lesions. Addison’s disease is primary adrenal failure, combined with hyperpigmentation in the oral cavity and diffuse hyperpigmentations in the nails [10]. The use of drugs that can cause hyperpigmentation, such as levodopa, tetracycline, ketoconazole, phenothiazine, oral contraceptives and anti-malarial drugs, must be considered in the differential diagnosis [11].
Chronic tobacco use is also known to cause similar lesions characterized by hyperpigmentations in the oral mucosa in particular [12].
In our case treatment with methotrexate and sulphasalazine was used, but the hyperpigmentations started before such treatment. What is important, the described patient did not have a smoking history. Peutz-Jeghers syndrome has autosomal dominant transfer and is seen at birth or in early childhood. It is characterized by intestinal polyposis and hyperpigmentations on the lower lip and in the oral cavity [13]. So this diagnosis seemed impossible in this patient.
Obviously the described patient was evaluated clinically, radiologically and with laboratory tests in respect of diseases with symptoms of hyperpigmentation and no particular cause of such changes was determined. There was a histopathological picture typical for LHS: homogeneous pigment changes, melanin deposition in basal keratinocytes, melanophage accumulation in the dermis, with normal size and normal count of the melanocytes and without the presence of atypical cells [14].
Laugier-Hunziker syndrome has a benign course apart from cosmetic problems and generally treatment is not necessary. Cryotherapy and laser therapy have been reported in some cases to be reliable and effective treatment options for LHS patients. Cryosurgery has also been used. Since relapses of lesions after treatment may occur, protection against sunlight is recommended as a method to reduce such a possibility [15].